全固態鋰電池中金屬鋰負極及其界面設計的研究進展
2021-08-12 10:16:56徐亞楠王克儉馬衍偉
材料工程 2021年8期
楊 杰,王 凱,徐亞楠,王克儉,馬衍偉
(1 中國科學院電工研究所,北京 100190;2 北京化工大學 機電工程學院,北京 100029)
在當前所有的能量轉換和存儲系統中,鋰二次電池是最為高效便捷的儲能器件,已廣泛應用在消費電子、航空航天、電動汽車和國防安全等領域。隨著科技的發展,新型電子器件/裝置對電池的長續航能力、高安全性等有著越來越高的要求。1991年,索尼公司開發出以炭材料為負極、含鋰化合物為正極的鋰離子電池,實現了鋰離子電池的商業化,其能量密度為80 Wh/kg;而當前鋰離子電池的能量密度已達到200~300 Wh/kg。目前,世界各國都在開發能量密度達到400~500 Wh/kg的二次電池,以滿足日益提升的能量密度需求。提升電池能量密度主要在于提升正、負極材料的比容量和正負極的電勢差。金屬鋰由于其高理論比容量(3860 mAh/g,比石墨負極高10倍)、低電勢(-3.040 Vvs標準氫電極)和低密度(0.59 g/cm3),被認為是理想的高能量密度鋰電池負極材料[1-3]。然而,在金屬鋰作為鋰電池負極與液態有機電解質組裝成金屬鋰電池使用時出現了一系列的問題,如金屬鋰極低電勢導致與有機電解液發生副反應,在金屬鋰表面形成固態電解質界面膜(SEI);充放電過程中鋰不斷溶出和沉積帶來大的體積變化,造成SEI膜不穩定,電解質不斷消耗;充電過程中鋰離子在金屬鋰表面的不均勻沉積導致產生鋰枝晶,鋰枝晶的不斷生長會逐漸刺穿隔膜造成電池短路,使得電池瞬間釋放大量能量。有機液態電解質本身的可燃性加上鋰枝晶短路帶來的能量釋放,導致金屬鋰電池容易引發火災爆炸等安全事故。當前雖然針對上述問題進行了大量的研究改進,但是其循環壽命和安全性仍然無法滿足要求[4]。
采用固態電解質(SSEs)取代易燃、易泄露的有機液態電解質,有望從根本上解決了金屬鋰電池(LMBs)的安全隱患[5]。固態電解質是一類在室溫或不太高的溫度下具有高離子電導率、低的電導活化能(<0.5 eV)和非常低的電子電導率的材料,又稱為快離子導體。固態電解質不存在泄漏問題、壽命長且易封裝,可在較高溫度和電壓下使用。采用金屬鋰負極、全固態電解質、正極組成的二次電池,稱為全固態金屬鋰電池(all-solid-state lithium metal batteries, ASS LMBs)。固態電解質(尤其是無機固態電解質)本身不易燃,Li+遷移數高,具有高的力學強度可以阻擋鋰枝晶生長,因此提升了電池的安全性[6];而且金屬鋰的使用提升了電池的能量密度,因此,ASS LMBs是業界認為的最有前景的下一代高能量密度、高安全性電池,是鋰電池發展的第二次里程碑。
鋰離子固態電解質主要包括無機固態電解質(氧化物、硫化物、硼氫化物和鹵化物等)、聚合物固態電解質(醚類、烷基類、有機硅等)以及復合電解質。無機固態電解質一般具有較高鋰離子導電性和寬電位窗口,日本東京工業大學Kanno教授課題組[7]在2011年開發出Li10GeP2S12硫化物電解質,離子電導率達到1.2×10-2S/cm,可以與液態電解液相媲美;最近,日本豐田汽車Kato等[8]開發的Li9.54Si1.74P1.44S11.7Cl0.3室溫下離子電導率高達2.5×10-2S/cm,甚至超過了大部分液態電解液。此外,無機固態電解質本身不具有可燃性,具有較高力學模量,可以有效阻擋鋰枝晶向正極的延伸,從而可以避免電池短路引起的起火爆炸等問題;但是無機固態電解質一般為陶瓷片、玻璃等材質,加工性能不好,難以與鋰電池規模化生產中的卷對卷工藝兼容。聚合物電解質具有本征柔性,可以采用現有鋰電池生產工藝,但是室溫下聚合物易結晶造成離子遷移受限,其室溫離子導電率較低(10-7~10-5S/cm)。有機-無機復合固態電解質(CPE)綜合了聚合物與無機電解質材料的優點,不僅具有良好的離子導電率(≤10-3S/cm),而且具有一定的力學強度和界面柔性,可以抑制鋰枝晶的穿透和減小界面電阻;但是徹底解決影響全固態鋰金屬電池長循環性和安全性的問題仍是一項挑戰[9]。
鋰枝晶是金屬鋰電池短路的主要原因,盡管固態電解質較高的力學強度可阻擋金屬鋰表面枝晶的生長,但無法阻擋鋰枝晶沿著固態電解質晶界、缺陷和孔洞處延伸生長[10-11]。當固體電解質(如LLZO和Li2S-P2S5)的電子電導率相對較高時,Li+會在電解質內部直接沉積,產生大量枝晶穿透電解質造成電池短路[12]。此外,采用固態電解質取代液態電解質,電極與電解質的界面由固液界面變為固固界面,增大了電荷穿過電極-電解質界面時的電阻,造成嚴重極化。另一方面,由于金屬鋰的電勢太低,會與固態電解質發生界面副反應,造成不穩定的SEI層。電極-電解質界面電阻和界面相容性也是影響鋰枝晶形成的重要因素。針對固態電池中金屬鋰枝晶生長以及鋰與固態電解質界面電阻大的問題,研究者們開發出多種解決方法,包括從金屬鋰的結構設計及表面改性、固態電解質界面修飾等[13-15]。本文主要總結全固態電池中金屬鋰負極及其界面設計的研究進展,包括如何阻止鋰枝晶生長、降低金屬鋰負極與固態電解質接觸電阻,提升界面相容性的研究策略。
1 鋰枝晶的生長原理與影響因素
大多數金屬沉積-溶解過程都無法完全可逆,鋰金屬在電池充放電循環過程中需要進行多次的沉積-溶解,因此會在金屬鋰負極表面形成大量枝狀結構鋰(鋰枝晶)。鋰枝晶產生的根源是不均勻成核,其生長原理如圖1所示。在電池充電時,Li+在電場作用下傳輸到負極界面,被來自外電路電子還原沉積在金屬鋰負極表面。但由于負極表面無法達到理想的平整性,因此電荷在負極表面凸起處大量聚集,電場強度大首先被快速沉積形成晶核。由于尖端放電效應,后續沉積大多發生在最初的晶核處;因此晶核逐漸生長形成枝狀鋰。在放電時,靠近負極界面的鋰枝晶電流密度較大,首先被溶解,造成根部斷裂,變為不可利用的“死鋰”,導致離子傳輸緩慢,容量降低,加劇了鋰枝晶的生長[16-18]。美國德州大學奧斯汀分校Goodenough課題組[19]從溫度和能量的角度,重點介紹了鋰枝晶有關熱力學影響因素:從能量角度,在較低的表面能和較高的遷移能下更容易形成鋰枝晶;從溫度角度,較高的溫度降低了表面遷移勢壘,促進了離子擴散,從而導致了較大沉積尺寸的低形核密度和更平滑的鋰沉積。所以可通過調節溫度或熱力學能來改變鋰的成核和生長行為,從而控制鋰枝晶的生長。清華大學張強課題組[20]研究表明鋰沉積在負極表面的最初形貌極大地影響了沉積的最終狀態,若前期沉積不均勻將導致局部枝晶生長過快,刺穿電解質導致電池短路。金屬鋰負極表面本身無序的凹凸不平會造成電場強度的局部不均勻分布,與固態電解質(特別是無機固態電解質)直接界面點接觸,會進一步導致Li+在金屬鋰負極的不均勻沉積和溶解,加劇鋰表面的枝晶生長和增大界面阻抗。枝晶的形核生長過程涉及化學、電化學、晶體學、分子動力學和熱物理學等領域,無法用單一的模型來說明全過程,有關枝晶生長的理論還在進一步研究中[17]。實際上,鋅、銅、鎳等很多金屬在電化學沉積時都會產生枝晶,對許多高電勢金屬的枝晶生長已探索出明確機理,但由于鋰金屬還原電位過低,枝晶的生長機理與其他金屬相差較大,目前尚未明確,因此枝晶問題也沒有得到徹底的解決[21]。
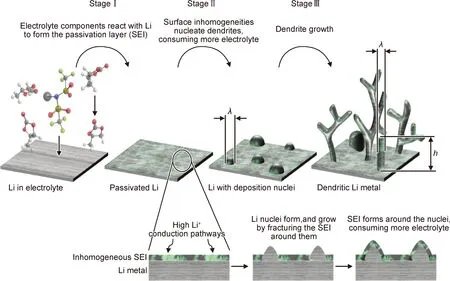
圖1 鋰枝晶生長原理圖[21]Fig.1 Structure illustration of Li dendrite growth[21]
現階段對鋰負極枝晶的研究多采用原位表征技術,比如原位透射電子顯微鏡(In-situTEM)[22-23]、原位掃描電子顯微鏡(In-situSEM)[24-25]等,這樣不需要拆解電池就可以觀察到在充放電過程中鋰枝晶生長的連續變化,對探究枝晶生長機理與影響因素提供了方便,從而有助于對金屬鋰電池的安全性和循環壽命的研究[26]。經過近40年的研究,研究者們提出了許多影響枝晶生長的因素,主要有:(1)電流密度:鋰金屬沉積-溶解過程中,電流密度對鋰枝晶的成核和生長產生了重要的影響[27]。Brissot等[28]提出的模型表示鋰枝晶生長的時間τ與電流密度J2成反比,在電流密度超過臨界電流密度(CCD)時會加速鋰枝晶的生長,而過低的電流密度充電時也會有少量枝晶出現。(2)金屬鋰體積變化:傳統鋰金屬電池用純鋰作負極,無骨架性,界面隨著電循環過程中電極材料體積膨脹產生巨大的內應力波動和變化,最終坍塌破裂和導致不均勻的鋰沉積,形成大量的鋰枝晶穿透電解質使電池失效。三維骨架的應用,促使鋰沉積在骨架的內孔中,有效地抑制了負極的體積變化和枝晶的產生[29-31]。(3)界面動力學:由于電極/電解質固固接觸或鋰與電解質副反應產生不穩定界面層(SEI),不利于鋰離子在界面的擴散和傳輸,且表面張力差,造成局部枝晶生長過快[32-34]。基于這些影響因素,研究者們對鋰金屬負極及鋰/電解質的界面開展了許多研究,如圖2所示:(1)鋰金屬負極:對金屬鋰表面做機械處理或引入親鋰性三維骨架,都減小了負極的體積變化,同時增大了負極比表面積,促使Li+在接觸面的均勻分布和降低局部電流密度,進而有效地抑制了枝晶的產生;引入可與鋰金屬合金化的保護層,增大界面浸潤性與穩定性,可顯著降低鋰/電解質界面電阻。(2)固態電解質:對固態電解質表面進行表面修飾,可達到與負極引入保護層類似的效果;采用無機/聚合物復合固態電解質,可以提高電解質的力學性能,進而抑制了枝晶的生長和死鋰的產生,聚合物的引入也增大了界面浸潤性,進而降低了金屬鋰與固態電解質界面電阻;采用原位生成聚合物電解質工藝,使得固態電解質與金屬鋰的界面接觸更緊密,促使Li+更均勻分布,也降低了界面阻抗[35-36]。
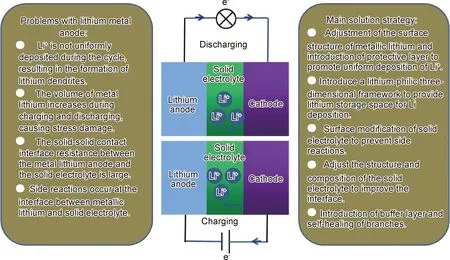
圖2 全固態鋰金屬電池充放電示意簡圖Fig.2 Schematic diagram of charging and discharging of all-solid-state lithium metal batteries
除此之外,還有許多其他直接或間接影響鋰枝晶生長的因素,如電解質中離子的濃度梯度和充電方式等。馬里蘭大學王春生團隊[37]組裝了Li/LLZO/Cu和Li/Li3PS4/Pt電池,使用原位中子深度剖析技術(NDP)在25,60 ℃和100 ℃下先后發現,LLZO和Li3PS4兩種電解質體相中Li+濃度不斷增大,鋰枝晶也變得更多。另外電池采用不同的充電方式也能產生不同的結果,浙江大學陸盈盈團隊[38]采用SEM分析了鋰金屬電極在恒流和脈沖電流經8次循環后表面和截面形貌,脈沖電流循環和恒流循環后的鋰金屬電極厚度分別增加了30 μm和84 μm,厚度增量更小;說明與直流充電相比,特定脈沖電流會改變枝晶的形核與生長動力學,電極表面的鋰枝晶生長可得到有效抑制,可以使電池壽命大幅度提高。由于本文主要總結全固態電池中金屬鋰電池的研究,因此未包含電解液濃度調控、電解液添加劑等液態電解質中金屬鋰保護的研究內容。
2 金屬鋰/固態電解質:金屬鋰的設計與修飾
2.1 金屬鋰表面微觀結構設計
金屬鋰負極表面無法達到絕對的平整,表面無序的凹凸不平會造成電場強度的局部不均勻分布,電場強度集中的地方優先成核-生長,從而造成了ASS LMBs在充電過程中形成大量鋰枝晶[39-40]。通過微納加工(MEMS)或者常規機加工等技術,設計制造具有周期性納微結構的圖案(比如納米孔陣列),降低局部電流密度,并引導電場線均勻分布,促進Li+在表面均勻分布。此外,這些納微結構設計還為鋰的沉積-溶出提供了一定的體積緩沖空間,從而緩解鋰枝晶的生長和負極材料體積膨脹[41]。Ryou等[42]通過納-微米針刺技術在金屬鋰表面輥壓制備了300 μm的深孔結構,使其擁有了更高的比表面積,在充放電電循環過程中具有局部降低的電流密度,比未處理的金屬鋰片倍率性提升了20%,循環穩定性提升了200%。SEM觀察到Li+沿孔洞側壁沉積,具有更小的界面阻抗,在不同的電流密度下與純鋰表面相比具有更少的枝晶生長和體積變化,如圖3所示。Park等[43]使用間隙控制型輥壓機對鋰金屬表面微機械沖壓得到了方形的點陣,通過SEM表征發現充電過程中孔內金屬鋰不產生鋰枝晶,溶解后孔徑可恢復至原來尺寸,結合COMSOL Multiphysics軟件有限元法模擬了Li在不同孔徑中的沉積-溶解過程,得出50 μm的最佳孔徑。
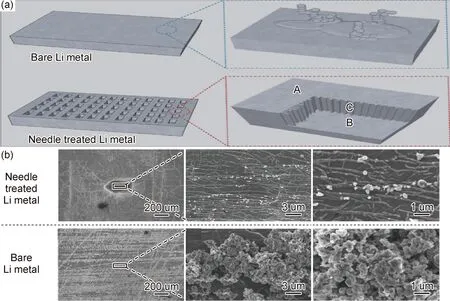
圖3 針刺處理前后鋰金屬表面及其鋰沉積后SEM圖[42] (a)鋰沉積在純金屬鋰表面和針刺處理過的金屬鋰表面圖;(b)在0.53 mA/cm2電流密度下沉積10 min后,純金屬鋰和針刺處理的金屬鋰表面SEM圖Fig.3 SEM images of the lithium metal surface before and after the needling process and after deposition of lithium[42](a)lithium deposited on the surface of pure metal lithium and surface image of the needle-treated metal lithium;(b)SEM image of the surface of pure metal lithium and the needle-treated metal lithium after being deposited for 10 minutes at a current density of 0.53 mA/cm2
2.2 三維(3D)骨架
除了鋰金屬表面結構設計外,研究者提出了引入三維骨架并用于負極的制備(或稱三維集流體)。制備時可將鋰通過熱澆注或電沉積法堆積到骨架中,促使負極合金化[44];還可以利用金屬鋰的延展性,通過冷壓方式直接將金屬鋰壓制到銅網、不銹鋼網等集流體中,或者將金屬鋰箔與鋁箔(或錫箔等)疊放到一起對折-壓制,重復多次形成鋰和鋁的層狀堆疊箔材。三維骨架結構能在充放電過程為鋰的溶出和沉積提供空間,有效緩沖鋰的體積變化;同時增大了負極比表面積,促使Li+在接觸面的均勻分布和降低局部電流密度,進而減少了鋰枝晶與電解質接觸而發生的副反應。圖4為充電過程中鋰沉積到銅片表面和三維銅骨架上循環沉積行為示意圖。三維骨架以導電框架為基礎,常見的有多孔金屬(如泡沫金屬銅和泡沫金屬鎳)、碳纖維薄膜、石墨烯薄膜等導電性框架;也有植物秸稈、干木、聚酰亞胺和玻璃纖維等非導電性框架,有天然的通道促使離子傳輸,在使用前常需要進行導電化(如炭化),以增強其導電能力,但因其力學性能較差和制備過程繁瑣限制了使用[45-48]。
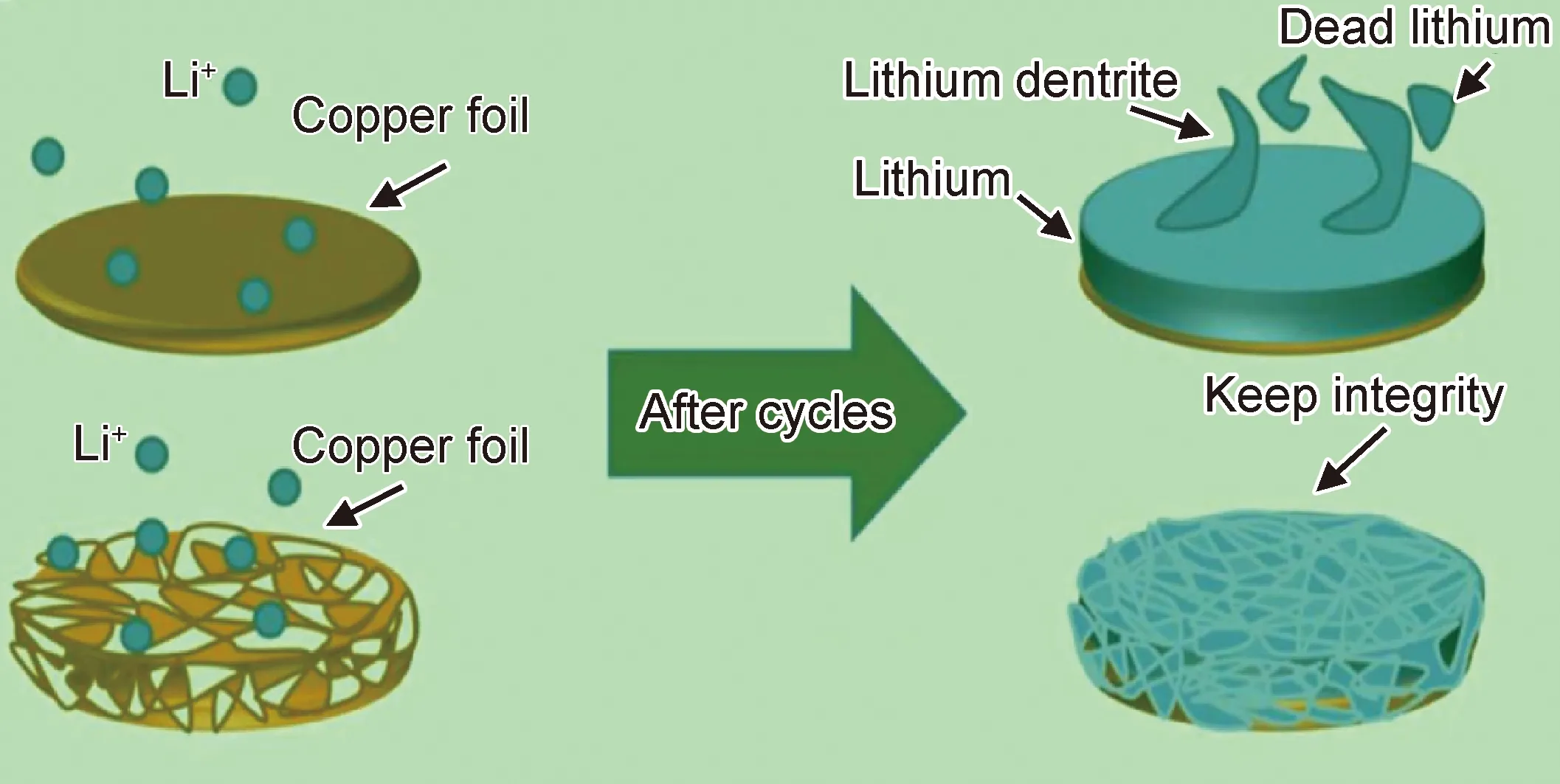
圖4 金屬鋰在二維銅箔及三維銅骨架上循環沉積行為示意圖[49]Fig.4 Schematic diagram of cyclic deposition behavior of lithium metal on two-dimensional copper foil and three-dimensional copper skeleton[49]
2.3 金屬鋰保護層
無機固態電解質(如石榴石型SSEs)一般親鋰性較差,當熔融金屬鋰與其表面直接接觸時,會產生較大的界面張力,不利于鋰離子在界面均勻分布和沉積,導致嚴重的枝晶生長和較大的界面電阻。研究者利用金屬鋰的強還原性,通過原位或非原位等方式在金屬鋰表面形成合金保護層,促進Li+均勻沉積,抑制枝晶的產生和生長[56-59]。此外,保護層一般是與鋰金屬合金化的材料,具有高離子電導率,因此可顯著降低界面電阻。
金屬鋰保護層的引入可以通過原位反應方法。例如,Goodenough課題組[60]使用具有高離子電導率的LiZr2(PO4)3與金屬鋰箔接觸反應原位生成了Li3P和Li8ZrPO4的鋰離子導電鈍化層,在80 ℃時,組裝的Li/LiZr2(PO4)3/Li電池(650 Ω·cm-2)具有比普通Li/石榴石SSEs/Li電池(1700 Ω·cm-2)大幅下降的界面電阻,也有效抑制了枝晶的生長。中科院化學所郭玉國課題組[61]開發出一種金屬鋰原位保護方法,將金屬鋰片直接浸入到多磷酸(PPA)物質的處理液中,原位生成Li3PO4界面層,不僅可以增強Li+在鋰金屬界面和電解質之間的傳輸,也可有效地抑制鋰枝晶生長和副反應的發生。Li3PO4的離子電導率較低(約10-9S/cm2),不利于Li+的快速均勻傳輸,影響了電池的循環性和倍率性,仍需進一步優化。北京理工大學黃佳琦團隊[62]采用化學液相反應法,在室溫下用氟化銅溶液與金屬鋰發生置換反應,在鋰金屬表面原位生成膜厚度約為2 μm的離子/電子混合導體界面層(MCI),Li+傳輸路徑如圖5所示,其具有優異的儲鋰性和高離子導電性,25 ℃時MCI的離子電導為1.79×10-4S/cm,電子電導為2.06×10-3S/cm,楊氏模量(12.9 GPa)遠高于傳統的SEI膜(0.63 GPa),完全達到了抑制鋰枝晶生長的要求值(6.0 GPa)。將具有MCI保護層的鋰金屬負極與LiNi0.5Co0.2Mn0.3O2(NCM)正極組裝成全電池,壽命可達400個周期,是純鋰金屬負極電池的8倍。在0.5 C倍率下,500周次循環后容量依然維持在50 mAh/g,相比于傳統鋰金屬界面的電池經過100次循環后容量降低到50 mAh/g,將電池的壽命延長了5倍,平均庫侖效率達到了99.8%,MCI膜有效地促進了均勻擴散和沉積,減少了鋰枝晶的生成。
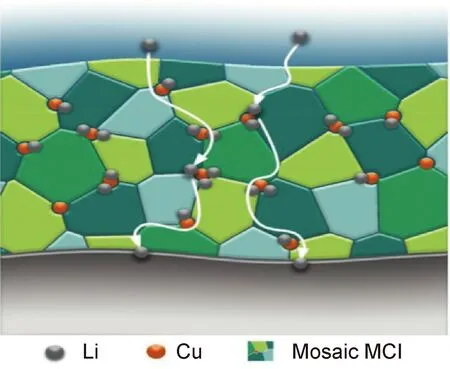
圖5 離子/電子導體混合導體MCI界面的Li+輸運示意圖[62]Fig.5 Schematic diagram of Li+ transport at the MCI interface of an ion/electronic conductor mixed conductor[62]
除了原位反應生成保護層外,還可以采用沉積的方式形成保護層。桑頓新能源在公開的專利[63]中報道了在惰性氣體中采用磁控濺射技術在金屬鋰片表面引入復合LiPON/LiF膜,與現有的單層Li3PO4界面保護層相比,該復合界面保護層具有更高的楊氏模量(LiF的剪切模量為55.1 GPa)和鋰離子電導率(約10-6~10-7S/cm2),且電化學穩定窗口更高(5 V以上)。采用Li2S和P2O5質量比為75∶25的混合物作電解質(LPS),將其組裝成Li@LiPON@LiF/LPS/TiS2全固態電池。在0.2 C下循環50周次后,放電容量和庫侖效率分別高達256.8 mAh/g和99.8%,有效地降低了界面電阻和抑制枝晶生長,增強了ASS LMBs循環性和倍率性。Li與Zn易發生合金反應,生成的合金有極高的離子擴散系數(4.7×10-8cm2/s)[64]。電子科技大學王志紅課題組[65]采用直流磁控濺射技術在鋰金屬片表面沉積Zn形成Li-Zn合金膜(厚度約600 nm),Li沉積前后表面形貌表征如圖6所示。使其與聚環氧乙烷(PEO)基聚合物電解質(電導率為1.28×10-5S/cm)組裝成對稱電池,在0.5 C倍率下循環可超過920 h(≈460周次),比未改性前壽命提升6倍以上,界面觀測無明顯枝晶生成。在組裝成Li-Zn/PEO/LiFePO4全固態電池后,相同倍率下進行測試,經過200周次循環后放電比容量為133.3 mAh/g,容量保持率為87.0%,平均庫侖效率達到了98.0%,各項性能相比純Li均有很大提高,從而說明引入Zn膜后的金屬鋰負極在循環過程中有更少的枝晶和死鋰產生。

圖6 原始鋰片與沉積鋰鋅合金后的光學照片與SEM形貌表征[65] (a)原始鋰片光學照片;(b)沉積鋅后的鋰片光學照片;(c)原始鋰片表面SEM圖;(d)部分包覆鋅的鋰片SEM圖;(e)沉積鋅的表面SEM圖;(f)沉積鋅的截面SEM圖Fig.6 Optical photographs and SEM morphology characterization of the original lithium sheet and the deposited lithium-zinc alloy[65](a)optical photo of the original lithium sheet;(b)optical photo of the lithium sheet after zinc deposition;(c)SEM image of surface of the original lithium sheet;(d)SEM image of lithium sheet partially coated with zinc;(e)surface SEM image of the zinc deposit;(f)cross-sectional SEM image of deposited zinc
3 金屬鋰/固態電解質:電解質設計與修飾
3.1 固態電解質表面修飾
2020年,加拿大西安大略大學孫學良和青島大學郭向欣教授課題組[72]采用酸刻蝕工藝,將致密的LLZTO陶瓷片浸泡在HCl溶液中1 h,原位蝕刻出厚度為30 μm的三維多孔結構的LLZTO(3D-LLZTO),并構建了未改性Li/LLZTO、親鋰性Li/LLZTO@ZnO和Li /3D-LLZTO@ZnO三種不同界面,鋰沉積行為如圖7所示,系統地探究了不同界面構造對鋰枝晶生長的影響。結果表明除了界面的潤濕性,循環過程中界面動力學穩定性同樣影響鋰枝晶的生長;組裝成Li/3D-LLZTO@ZnO/Li對稱電池,在0.5 mA·cm-2電流密度下可實現600 h以上的穩定循環,但在未改性Li/LLZTO和Li /LLZTO@ZnO鋰對稱電池中可清楚地觀察到鋰枝晶的生長。3D-LLZTO@ZnO界面能有效降低局部電流密度,減小Li體積變化,促進Li與LLZTO緊密接觸,有效抑制鋰枝晶的生成。2020年,浙江工業大學陶永新教授課題組[73]通過引入Li2S添加劑對PEO-LiTFSI進行界面改性,成功制備了PEO-LiTFSI-Li2S固態電解質,其界面阻抗(374 Ω)低于PEO-LiTFSI(557 Ω);結合低溫透射電鏡(Cryo-TEM)和分子動力學模擬表明,Li2S的引入加速了LiTFSI的分解,可原位構筑富含高傳導LiF的界面,生成的LiF可有效抑制PEO鏈中C—O鍵的斷裂,從而阻止Li和PEO的持續副反應,使Li/PEO界面在循環過程中保持穩定;組裝成Li/PEO-LiTFSI-Li2S/LiFePO4電池,比容量為140 mAh/g,在50 ℃和0.5 C倍率下,庫侖效率高達99%,循環1000周次后容量保持率為85%,明顯優于PEO-LiTFSI的電池(77%, 150周次),所以富含LiF界面的ASS LMBs結構中表現出更好的循環能力和穩定性,有效降低了界面電阻和抑制鋰枝晶的生長。
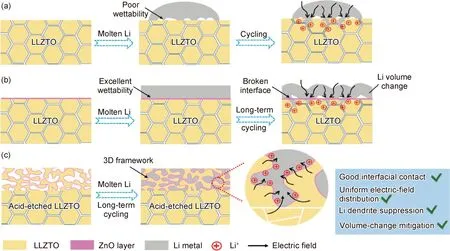
圖7 三種不同界面鋰沉積行為示意圖[72](a)LLZTO/Li界面;(b)LLZTO@ZnO/Li界面;(c)3D-LLZTO@ZnO/Li界面Fig.7 Three different interfacial behavior schematic of lithium deposition[72](a)LLZTO/Li interface;(b)LLZTO@ZnO/Li interface;(c)3D-LLZTO@ZnO/Li interface
3.2 固態電解質復合
無機固態電解質具有優異的室溫離子電導率和力學性能,但脆性大,加工成型過程復雜,且與金屬鋰固固接觸界面電阻大,不利于Li+均勻沉積;聚合物電解質(SPE)具有優異的柔韌性和可加工性,可實現與金屬鋰接觸緊密,但室溫下離子電導率較低,需要在較高溫度下使用,如PEO室溫下離子電導率僅10-6~10-7S/cm,需要在60 ℃以上的溫度下才能工作[74]。為了發揮不同電解質的優點,研究者開發出有機-無機復合電解質。無機粒子與聚合物電解質相互作用不僅可以減小聚合物的結晶,提升室溫離子導電率;還可以提高聚合物的力學強度,進而抑制了枝晶的生長和死鋰的產生;聚合物的引入也降低了金屬鋰與固態電解質界面電阻,其相互作用機理如圖8所示。2016年,中科院寧波材料所許曉雄研究員課題組[75]將具有超高Li+電導率的三元硫化物Li10GeP2S12(LGPS)與聚環氧乙烷(PEO)鋰鹽體系進行復合,制備出復合固體薄膜電解質(SPE)。該電解質柔性可彎曲且膜厚可控,室溫下具有較高Li+電導率(1.18×10-5S/cm),電化學窗口達到5.7 V。組裝成全固態Li/SPE/LiFePO4電池,在60 ℃和0.5 C倍率下循環50周次后電池比容量高達92.5%,有效地抑制了鋰枝晶生成。同年,該課題組[76]系統地研究了固體增塑劑丁二腈(SN)對PEO基電解質離子電導率的影響,采用傳統的溶液澆鑄法成功地制備了一種新型固體聚合物電解質(PEO18-LiTFSI-1%LGPS-10%SN),其在25 ℃時最大離子電導率為9.1×10-5S/cm,比PEO-LiTFSI的離子電導率提高了15倍。組裝成Li/PEO18-LiTFSI-1%LGPS-10%SN/ LiFePO4電池,在40 ℃以下具有良好的循環性和倍率性,在0.5 C倍率時最大放電比容量為138.4 mAh/g,甚至在循環100周次后容量保持率接近90%。這種新型SPE具有優異的電化學性能,有效抑制了枝晶的生長。除此之外,該課題組[77]采用不同粒徑和質量比的Li1.5Al0.5Ge1.5(PO4)3(LAGP)活性顆粒填充到PEO中,制備了不同類型的復合電解質(PEO-LAGP),結果表明對于相同含量的LAGP,粒徑越小,PEO-LAGP離子導電率越高。最小粒徑LAGP-I在含量為20%(質量分數,下同)時電解質具有最高電導率(60 ℃時為6.76×10-4S/cm)。組裝成Li/PEO-20%LAGP-I/LiFePO4電池,在60 ℃時1 C倍率下循環50周次后容量為100 mAh/g(容量保持率近90%)。相同情況下的Li/PEO/LiFePO4電池,循環50周次后容量保持率僅為44%,所以在PEO加入LAGP-I粒子,使其具有良好的界面穩定性和與電極的相容性,有效地抑制了鋰枝晶的生長。中科院北京納米能源所孫春文課題組[78]報道了由Li7La3Zr2O12(LLZO)顆粒和PVDF-HFP聚合物基體形成的復合固態電解質(CSE),并采用流延法制備了CSE膜。在Li/CSE/Li對稱電池中測試CSE對鋰的穩定性,連續工作超過400 h而無短路,能有效抑制鋰枝晶的生長。Choi等[79]利用離子液體陰離子的吸附和固化作用,將三氟甲基磺酰亞胺鋰(LiTFSI)、離子液體(Pyr14TFSI)和納米BaTiO3顆粒以一定比例混合,開發了一種不含聚合物的新型復合固體電解質(CSE),30 ℃時離子電導率為1.3×10-3S/cm,Li+遷移數為0.35。組裝成Li/CSE/LiCoO2和Li/CSE/LiFePO4電池,均表現出良好的循環性與安全性,有效促進Li+均勻分布,抑制了鋰枝晶的生長。
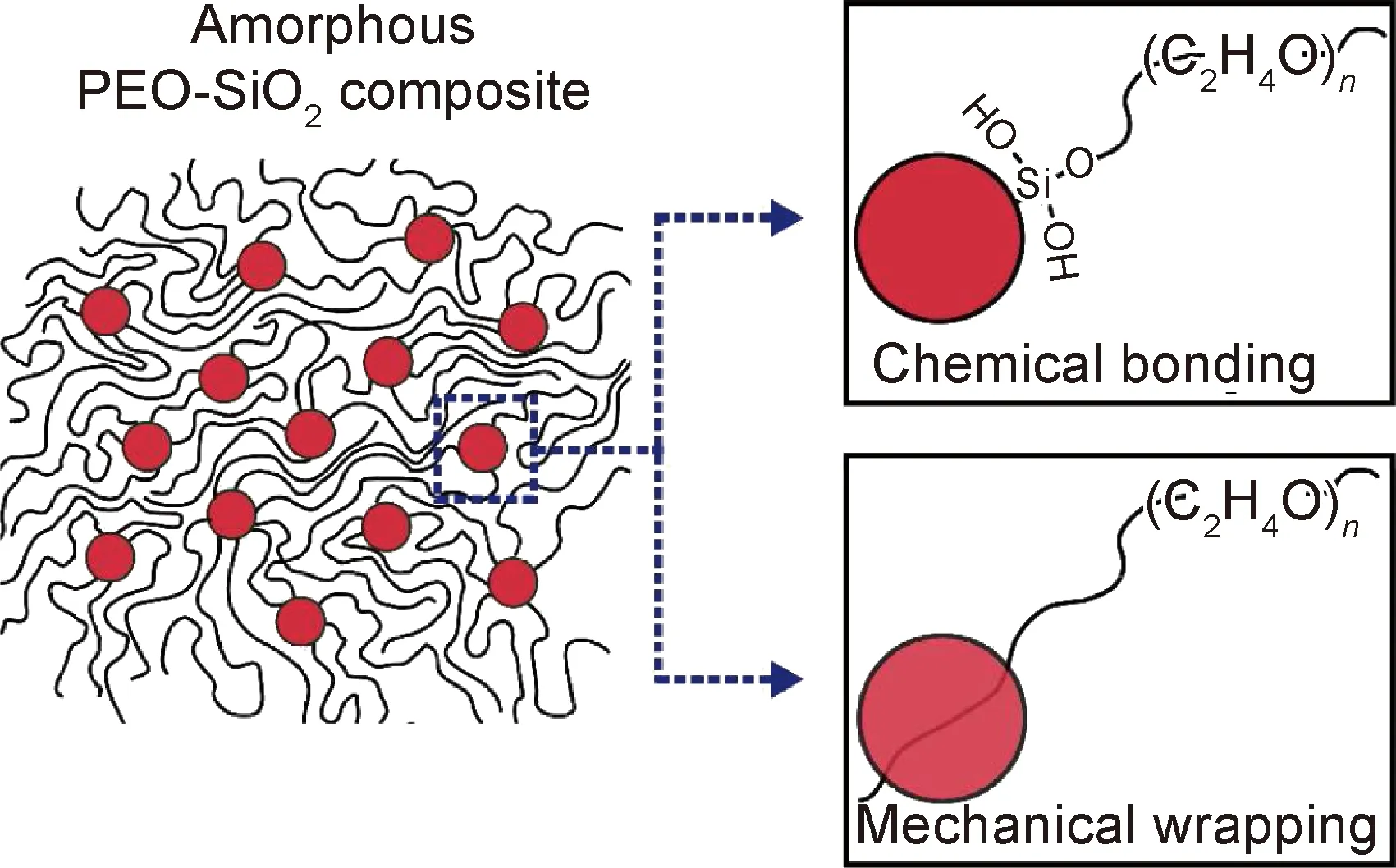
圖8 有機-無機復合電解質的兩種相互作用機理[80]Fig.8 Two interaction mechanisms of organic-inorganic composite electrolyte[80]
除了無機粒子與聚合物共混形成的復合電解質,研究者為了針對性地解決正負極界面不同需要,開發設計了層狀結構的復合電解質[81-82]。宇通集團[83]設計了在厚度方向上有濃度梯度的三層結構Li6.75La3Zr1.75Ta0.25O12(LLZTO)/PEO復合電解質(HSE),從正極側到負極側的各電解質層中,厚度依次為5,10,5 μm,其中LLZTO含量分別為95%,50%,5%。正極側LLZTO含量高可以耐受更高的電壓,提供更高的離子電導率,負極側PEO含量高可更好地降低了界面電阻,也緩解了金屬鋰體積膨脹。在1 C倍率下進行Li/HSE/Li對稱電池測試,循環950周次后容量剩余80%。相同條件下,LLZTO與PEO質量分數各為50%的普通共混復合電解質,循環560周次后容量僅剩余80%。Goodenough課題組[84]制備了聚合物/陶瓷/聚合物三明治結構復合電解質(PCPSE),其中聚合物是聚甲基丙烯酸甲酯(PMMA),而陶瓷采用Li3AlO3Ti7(PO4)3(LATP)電解質。在這種電解質結構設計中,無機層阻擋了鋰鹽陰離子的輸運,減小了金屬鋰與電解質界面空間電荷的影響,降低界面阻抗。由這種電解質PCPSE組裝成Li/PCPSE /LiFePO4電池,在0.51 mA/cm2的電流密度和2.5~4.0 V下充放電640周次,電池比容量約為100 mAh/g,庫侖效率為99%,顯示了優異的性能。美國馬里蘭大學Hu課題組[85]利用石榴石型的Li6.4La3Zr2Al0.2O12(LLZAO)納米纖維制備了三維骨架,填充聚合物PEO后形成復合固態電解質,如圖9所示。在這種三維離子導體結構中,鋰離子可以沿著LLZAO傳遞,形成快速離子傳輸通道,電解質的室溫Li+電導率達到 2.5×10-4S/cm。此外,與普通聚合物電解質相比,三維LLZAO網絡骨架增強了力學性能,可更有效地抑制鋰枝晶的生長。
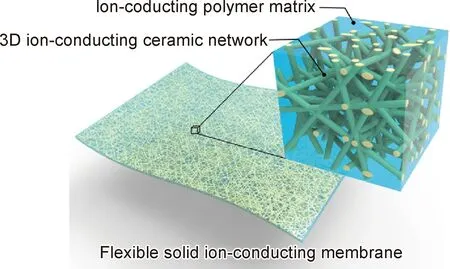
圖9 以三維LLZAO納米纖維為骨架制備的PEO基有機無機復合電解質[85]Fig.9 PEO-based organic-inorganic composite electrolyte prepared with three-dimensional LLZAO nanofibers as the framework[85]
通過將不同分子量或者含有不同官能團的聚合物進行共混,也開發出了性能優異的復合電解質。2016年,中科院寧波材料所許曉雄課題組[86]采用一鍋法將三羥甲基丙烷三縮水甘油醚(TMPEG)與聚乙二醇二胺(NPEG)交聯改性,成功地制備了一類具有可調網絡結構的共聚物電解質(SNPE)。通過改變TMPEG與NPEG的摩爾比或NPEG的分子量可以調節SNPE網絡結構。研究發現,當TMPEG中環氧基與NPEG中胺基的摩爾比為1∶1和EO/Li+摩爾比為16∶1時的網絡結構為TMPEG-NPEG4K[2∶1]-16∶1,在30 ℃時其最大離子電導率1.1×10-4S/cm,比PEO基電解質的離子電導率提高了18倍。將其組裝成Li/SNPE/LiFeO4電池,在60 ℃和1 C倍率下循環100周次后,比容量為132.7 mAh/g,容量保持率高達90.5%。相比之下的PEO-16∶1 SPE電池,循環5周次后比容量開始急劇衰減,50周次后容量保持僅為6.7%,表明SNPE與Li具有良好的相容性,可有效抑制鋰枝晶的生長。2019年,西安交通大學宋江選課題組[87]將PVDF-HFP和PEO作為混合基體,并摻入快離子導體Li1.5Al0.5Ge1.5(PO4)3(LAGP)和溶劑化離子液體LiFSI-TEGDME(SIL)作為增塑劑,成功制備了高性能的復合聚合物電解質(PPLS),制備過程如圖10所示。PPLS室溫下具有離子電導率高達3.72 mS/cm、優良的機械強度(16 MPa)和穩定的寬化學窗口(4.9 V);組裝成Li/PPLS/LiFePO4電池,在室溫0.1 C的電流密度下循環100周次后電池比容量高達158 mAh/g,所以該新型固態聚合物電解質有效改善了鋰金屬負極界面穩定性和抑制了鋰枝晶的生長。2020年,華南理工大學劉軍教授課題組[88]通過靜電紡絲制備了PVDF-HFP/聚丙烯腈(PAN)/PVDF-HFP三明治結構的納米纖維膜。以此納米纖維膜作為骨架,以聚己內酯二醇(PCL)為聚合物基體,引入磷酸三甲酯(TMP)作增塑劑來降低聚合物基體的結晶度,制備了復合型固態電解質膜(PPT-CPE),制備過程如圖11所示。組裝成Li/PPT-CPE/LiFePO4全固態電池,該電池在30 ℃和0.5 C電流密度下初放電容量為131 mAh/g,15周次充放電循環后增加到142 mAh/g,并保持在該值附近,400周次循環后電池容量保持率在80%左右,具有較高的比容量和優異的循環穩定性,該結果表明PPT-CPE聚合物電解質具有良好的鋰枝晶抑制能力。
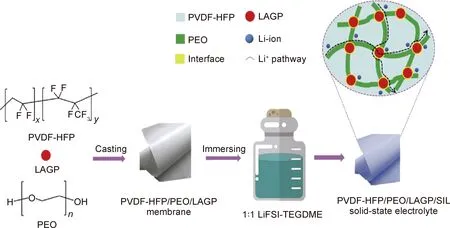
圖10 PPLS(PVDF-HFP/PEO/LAGP/SIL)固態電解質的制備工藝[87]Fig.10 Fabrication process of the PPLS (PVDF-HFP/PEO/LAGP/SIL) solid-state electrolyte[87]
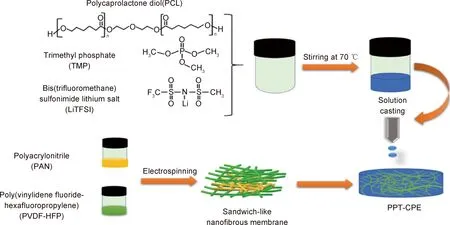
圖11 三明治狀納米纖維骨架及PPT-CPE復合聚合物電解質制備過圖[88]Fig.11 Preparation of sandwich nanofiber framework and PPT-CPE composite polymer electrolyte[88]
此外,研究者也通過電解質官能團修飾對固態電解質進行改性。許多氫鍵型自修復材料如超分子橡膠(SR)、脲基嘧啶酮(UPy)聚合物、羧酸化聚氨酯(CPU)和聚丙烯酸(PAA)等,具有優異的化學和電化學穩定性,以及較高的自修復效率,因此被用于鋰電池電解質和電極材料中,來提高電池的安全性和長循環性[89]。2018年,上海科技大學劉巍課題組[90]提出一種含脲基的自修復復合固態電解質,包括自修復聚合物和粒徑為500 nm無機固態電解質顆粒(Ga0.25Li6.25La3Zr2O12),其機理是脲基通過氫鍵結合形成交聯結構,材料破壞后將斷面接觸在一起,可以利用氫鍵間的相互作用實現材料自修復。這種電解質具有優異的柔性和力學性能,可有效抑制鋰枝晶的生長。組裝成Li/復合電解質/Li對稱電池,在20 mA/cm2下循環1500周次,未觀察到明顯電壓波動,電解質自修復功能大大地提高了電池的使用壽命。清華大學張強課題組[91]提出一種陰離子固定的聚合物基(PEO)、陶瓷(LLZTO)、鋰鹽(LiTFSI)組成的復合固態電解質(PLL)來抑制鋰枝晶生長。PLL固態電解質具有高達5.5 V的電壓窗口,鋰鹽的陰離子TFSI-被聚合物基體和陶瓷填料束縛,可誘導空間電荷和Li+的均勻分布,從而形成無枝晶鋰沉積。組裝成Li/PLL/LiFePO4全固態電池,在0.1 C倍率下,比容量約為155 mAh/g,庫侖效率為99%。在100周次電循環后容量衰減率達13%,保持了良好的循環性能。
3.3 原位生成固態電解質
為了提升固態電解質與金屬鋰的界面接觸,研究者開發出在電池內部原位生成的固態電解質。原位生成聚合物電解質減少了聚合物溶解和干燥成膜等復雜工序,使得電解質的制備和電池的組裝能同時進行,其原理是將一種或多種聚合物單體、引發劑(部分聚合不需要引發劑)和鋰鹽等按一定比例混合均勻后組裝電池,在一定的條件(如熱引發、伽馬射線等)下引發聚合反應。根據聚合原理不同,原位聚合包括自由基聚合、陰離子聚合、陽離子聚合、凝膠因子引發聚合和無引發劑聚合等方式,可根據聚合物單體本身特性選擇性使用[92]。原位生成電解質工藝截面簡圖和優缺點如圖12所示。
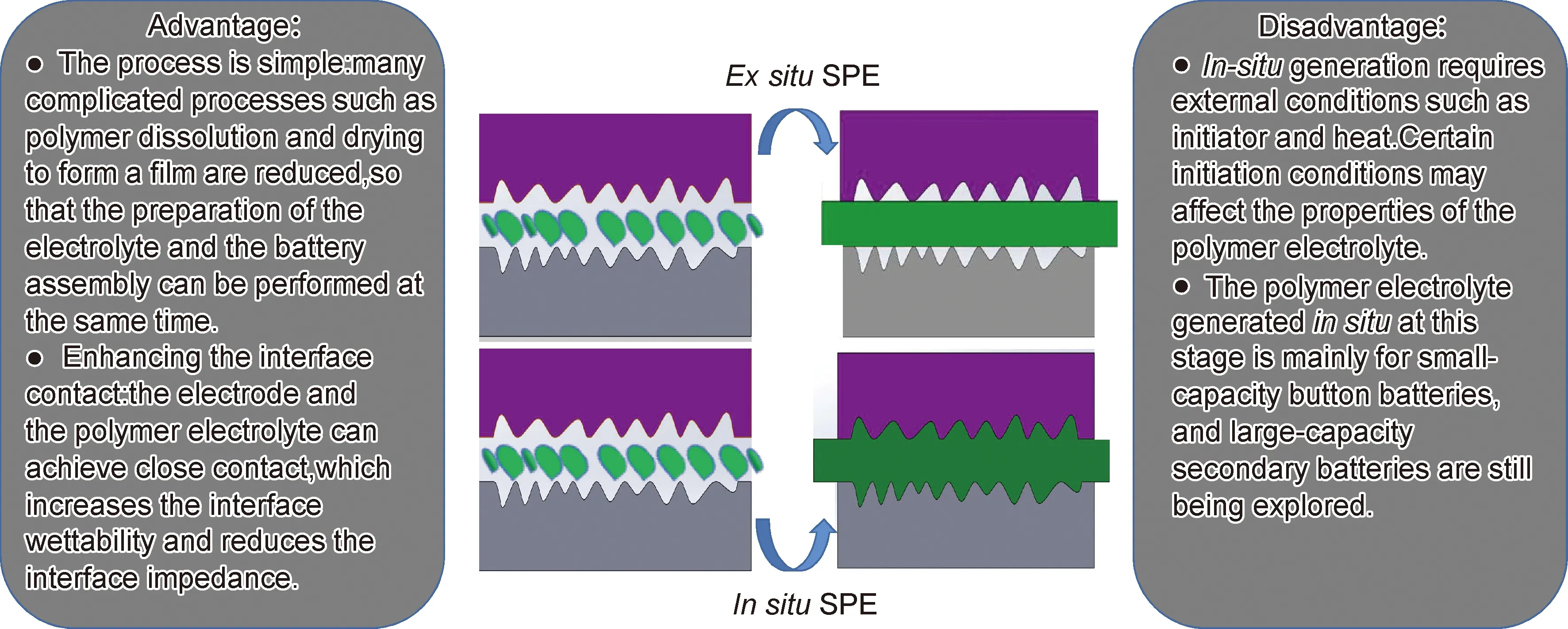
圖12 傳統與原位生成聚合物電解質工藝截面簡圖和原位生成聚合物電解質工藝原位優缺點Fig.12 Cross-section diagrams of traditional and in-situ polymer electrolyte process and their advantages and disadvantages
2017年,中科院青島能源所崔光磊課題組[93]利用碳酸亞乙烯酯(VC)的自由基聚合反應,以偶氮二異丁腈(AIBN)為引發劑,在電池內部原位生成聚碳酸亞乙烯酯/雙氟草酸硼酸鋰(LiDFOB)固態聚合物電解質(PVCA-SPE)。其在50 ℃時離子電導率為9.82×10-5S/cm。原位生成的PVCA-SPE與金屬鋰界面具有良好的相容性,組裝成Li/PVCA-SPE/Li對稱電池,經過600 h的恒電流充放電測試后未出現明顯枝晶現象,如圖13所示。不僅是純聚合物電解質可以原位生成,2018年該課題組通過原位聚合方法制備了PVCA/LiDFOB/ LSnPS有機-無機復合固態電解質。其溫離子電導率為2×10-4S/cm,電化學窗口大于4.5 V。制備成LiFe0.2Mn0.8PO4/PVCA-LSnPS/Li電池,在0.5 C倍率下循環140周次容量為116 mAh/g(初始為130 mAh/g),庫侖效率在99%以上[94]。中電第十八研究所趙冬梅課題組[95]將LLZO粉末與聚偏氟乙烯-六氟丙烯(PVDF-HFP)膠體液按一定比例混合,采用濕法涂覆的方式制備了0.05 mm的功能性離子導體陶瓷復合隔膜,并以鋰金屬為負極和NCM11為正極組成電芯。然后將電芯置于裝有電解液前驅體的不銹鋼殼體或鋁塑膜中一定壓力下加熱使電解液發生熱聚合,真空封裝后得到原位聚合固態電池。在2.5~4.2 V的電壓下,對該電池在0.1 C的倍率進行充放電測試,首周放電比容量為181.1 mAh/g,
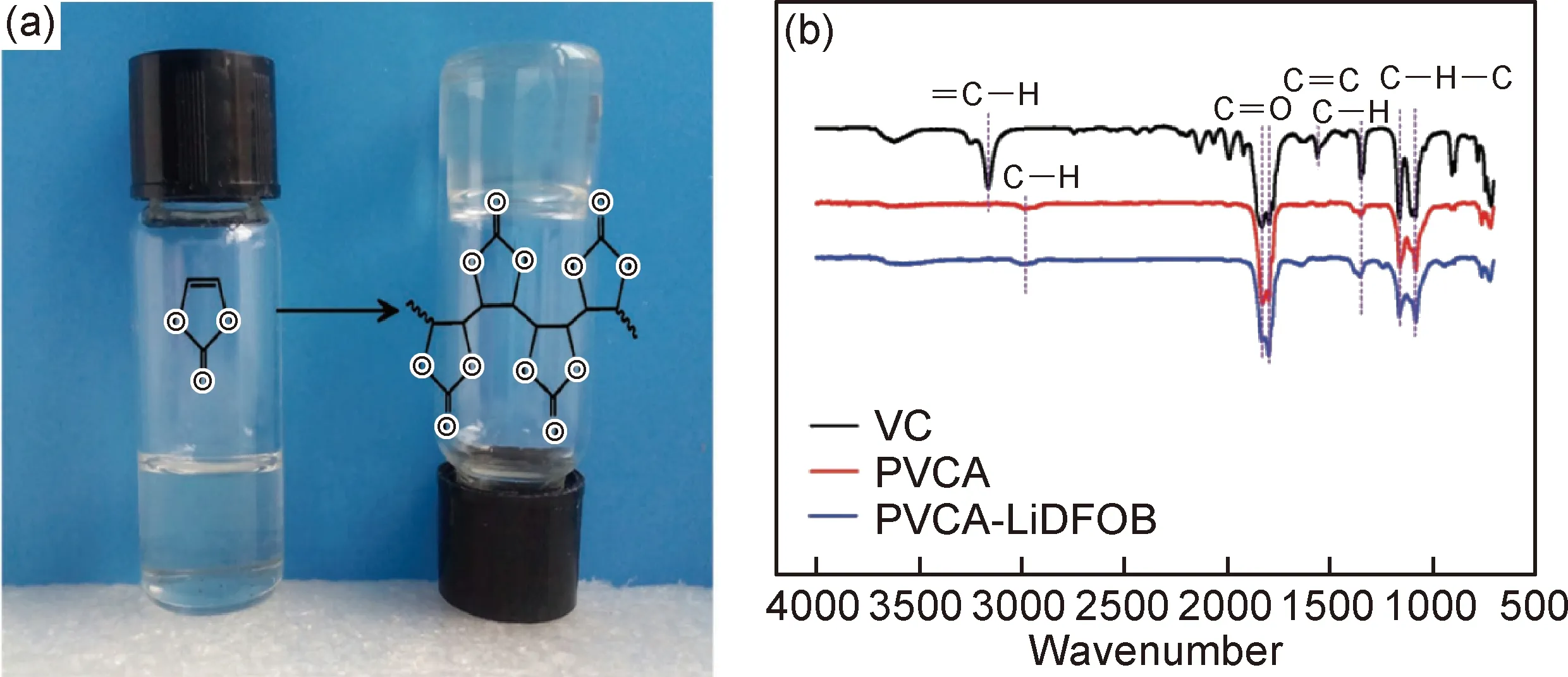
圖13 VC在60 ℃加熱24 h后原位聚合成PVCA的圖像(a)和VC,PVCA和PVCA-LiDFOB的FTIR光譜比較(b)[93]Fig.13 Typical image of in situ polymerization of VC into PVCA after heating at 60 ℃ for 24 h (a) and FTIR spectra comparison of VC, PVCA and PVCA-LiDFOB(b)[93]
循環100周次后,放電比容量保持為96.5%,平均庫侖效率為99.7%。此原位制備方式有效抑制了鋰枝晶生長,提升了固態電池倍率性與循環壽命。
4 其他研究策略
除了上述在對金屬鋰和固態電解質的設計和修飾外,也有許多其他方法(單離子導體、加壓等)用來克服鋰枝晶生長和降低界面電阻。在傳統聚合物電解質中,鋰鹽的陰、陽離子均可自由遷移,這會造成不均勻的極化電場,在界面處形成空間電荷,促使鋰枝晶的生成[96]。通過簡單的靜電相互作用或共價鍵將陰離子固定在聚合物基體中,形成單鋰離子導電聚合物電解質(SLICPEs)。2020年,中南大學劉洪濤課題組[97]成功合成了一種含氰基(—C≡N)官能團的新型單鋰離子導電聚合物鋰鹽——聚[(氰基)(4-苯乙烯磺酰亞胺)鋰](LiPCSI),不僅可以分散陰離子中心的負電荷,還可以提高離域陰離子的電化學穩定性,有效地限制了陰離子的移動。合成的PEO8-LiPCSI電解質在300 ℃的高溫下不發生分解,具有高達5.53 V(vsLi+/Li)的電化學穩定窗口,同時發現LiPCSI可以有效破壞PEO鏈段的規整度,降低了SPE的結晶度,具有高的Li+電導率(Li+遷移數0.84)。組裝成Li/PEO8-LiPCSI/LiFePO4電池,在0.1 C倍率下首周放電比容量為141 mAh/g,在循環80周次后容量保持率在85%以上,有效地抑制了SPE雙離子導體帶來的極化及鋰枝晶生長問題。
此外,對電池在制備和組裝過程中的施加一定壓力,也可讓金屬鋰與電解質界面接觸良好[37]。美國加州大學圣地亞哥分校Meng課題組[98]研究了Li/Li6PS5Cl/Li對稱電池在5 MPa下,電池可穩定地沉積-剝離1000 h以上,通過X射線斷層掃描發現在高壓力下電池循環過程中電解質會出現低密度轉化趨勢,相比無壓力的對稱電池,有效地抑制了枝晶生長和降低界面電阻。2020年,美國戴頓大學Wang團隊[99]研究了外部壓力對硫化物SSE電池Li金屬負極在沉積和溶解Li過程的影響,研究發現對Li溶解所需壓力與溶解電流密度呈正相關,對于鋰沉積較高的壓力導致較低的最大允許電流密度(超過該電流密度將產生鋰枝晶);同時證明了在高速溶解Li時需要較高的外部壓力,而低于4.5 MPa的外部壓力可在電流密度0.6 mA/cm2下沉積3 mAh/cm2的Li。所以外部壓力Li的沉積與溶解影響是很大的,通過有效的控制外部壓力可抑制鋰枝晶生長和延長電池壽命。
5 結論與展望
鋰金屬作為全固態鋰二次電池理想的負極材料,是實現能量密度大于400 Wh/kg的關鍵。本文綜述了近年來全固態電池中克服負極鋰枝晶和固固接觸界面電阻大的研究策略。
(1)對金屬鋰表面機械處理形成大量的周期性的納-微結構圖案以及引入三維骨架,實現降低局部電流密度,引導Li+沉積,為Li+在沉積-溶出過程的體積變化提供緩沖空間,有效地抑制了鋰枝晶的生長和負極體積膨脹。
(2)采用原位或非原位的方法在金屬鋰表面或者無機固態電解質表面引入保護層,引導Li+均勻沉積,改善了無機固態電解質親鋰性差和固固接觸濕潤性差的缺陷,抑制了枝晶的生長和降低界面電阻。
(3)采用復合固態電解質,無機粒子與聚合物電解質(或者聚合物與聚合物、有機小分子與聚合物)優勢互補,不僅可以減小聚合物的結晶,提升室溫離子導電率,還可以提高聚合物的力學強度,進而抑制了枝晶的生長和死鋰的產生。
(4)在電池內部原位生成固態電解質,不但有效提升了固態電解質與金屬鋰的界面接觸,降低界面電阻和抑制枝晶生長;還可減少聚合物溶解和干燥成膜等復雜工序,使得電解質的制備和電池的組裝能同時進行,有利于固態電池的批量制備。
(5)制備單離子導體、減少空間電荷效應影響;從制備工藝(加壓、調控鋰沉積電流)出發,改進電解質/電極的界面接觸,引導鋰的成核與生長,從而減少鋰枝晶生長,提升界面相容性。
目前對ASS LMBs研究仍處于初始階段,許多研究者對鋰負極、電解質的設計以及兩者的界面修飾進行了大量的研究,很大程度地完善了ASS LMBs的綜合性能,對抑制鋰枝晶的生長和降低界面電阻取得了很大突破,但并沒有徹底解決這些問題,目前仍不能滿足長續航能力和商業的實際需求。在該領域未來仍然要面對多方面挑戰。
(1)機理:當前,鋰枝晶的形成與具體電解質體系相關,仍然缺乏十分準確的普適性模型。需要在晶體熱力學、動力學等角度繼續探索,采用多尺度高分辨先進表征技術,結合分子動力學和熱力學理論計算,對鋰枝晶模型開展進一步研究。
(2)界面:金屬鋰與固態電解質的界面構筑將很大程度上影響電池的最終性能。相較于傳統的固液界面,固固界面的界面電阻較大,造成電池極化、影響容量和倍率;界面點接觸以及界面副反應產物,都會導致電場分布不均勻,影響Li+在金屬鋰負極的均勻沉積和溶解,造成枝晶生長,影響電池的庫侖效率和壽命。雖然,當前在界面構筑方面學術界已經開展了諸多研究,并取得了長足進展。但是未來仍然需要通過對鋰金屬負極和電解質界面的進行原子、分子尺度的研究,開發簡潔的界面構筑和修飾技術,實現完美、穩定的界面構筑。
(3)制備:在電池制備方面,需要開發新型制備技術,將固態電解質制備和正負極構建相結合,獲得工藝簡單、能規模化的制備工藝。此外,還需要在電池器件結構、疊片和封裝上進行創新性設計,獲得適合金屬鋰電極的制備技術。
猜你喜歡
小讀者(2021年2期)2021-03-29 05:03:48
當代陜西(2020年13期)2020-08-24 08:22:02
瘋狂英語·新悅讀(2019年11期)2019-12-18 05:14:16
華人時刊(2019年13期)2019-11-17 14:59:54
NBA特刊(2018年21期)2018-11-24 02:48:04
文苑(2018年22期)2018-11-19 02:54:14
制造技術與機床(2017年5期)2018-01-19 02:49:17
金秋(2017年4期)2017-06-07 08:22:16
中國材料進展(2016年10期)2016-12-26 06:50:20
濰坊學院學報(2016年2期)2016-12-01 13:00:11
