炎性巨噬細胞糖酵解水平與動脈粥樣硬化的研究進展*
2021-09-01 08:30:54曹小如趙雪竹譚凡成
中國病理生理雜志 2021年8期
關鍵詞:小鼠
曹小如, 趙雪竹, 譚凡成, 田 野, 時 颯△
(1哈爾濱醫科大學病理生理學教研室,黑龍江哈爾濱150001;2哈爾濱醫科大學附屬第一醫院心血管內科,黑龍江哈爾濱150001)
動脈粥樣硬化(atherosclerosis,AS)作為一種慢性代謝性疾病,是冠心病、腦卒中和外周血管疾病的共同病理學基礎,常伴有動脈炎癥反應[1]。根據AS的“炎癥學說”,在早期血管病變中,循環中的單核細胞募集至病變部位,浸潤至血管壁內膜下并分化形成巨噬細胞,分泌細胞因子介導免疫清除反應[2]。隨著斑塊進展,病原體相關分子模式[如脂多糖(li?popolysaccharides,LPS)]或損傷相關分子模式[如氧化型低密度脂蛋白(oxidized low-density lipoprotein,oxLDL)]可通過與巨噬細胞膜表面模式識別受體結合,激活斑塊內巨噬細胞炎癥反應,加劇AS發展[3]。近年,代謝活動參與調節免疫細胞炎癥功能備受關注[4]。Bekkering等[5]報道,與正常對照組相比,有缺血癥狀AS患者循環中的單核細胞糖酵解代謝明顯增加,強調了糖酵解過程參與AS進展的臨床意義。斑塊內巨噬細胞糖酵解代謝增加表現出炎性M1型巨噬細胞表型,而降低糖酵解可以減輕巨噬細胞炎癥,進而抑制AS進展[6-8]。因此,本文綜述炎性巨噬細胞的糖酵解及其調控機制,以及巨噬細胞糖酵解代謝調控AS相關研究,并探討降低炎性巨噬細胞糖酵解減輕AS的治療策略。
1 細胞糖酵解代謝增加的意義
1923 年,奧托·沃伯格證實腫瘤細胞在有氧條件下仍以較高速率的糖酵解代謝為主,這一現象被稱為Warburg效應[9]。進一步研究證實,由于惡性增殖的腫瘤細胞對生物大分子和生物能量的需求增加,細胞糖酵解代謝增加,從而提高自身對缺氧環境的耐受能力,以及在有氧條件下與正常細胞競爭生存的能力。高通量糖酵解在短時間內可以比線粒體氧化磷酸化產生更多ATP[10]。并且,糖酵解中間產物6-磷酸葡萄糖分流到磷酸戊糖途徑(pentose phos?phate pathway,PPP)生成核糖、NADPH等促進核酸、脂肪酸和固醇等大分子物質合成,從而參與細胞核、膜結構生成和氧化還原反應調控,促進細胞合成代謝[11]。此外,一般認為丙酮酸進入線粒體生成乙酰輔酶A,作為三羧酸(tricarboxylic acid,TCA)循環的主要碳源。而Hui等[12]證實在缺氧或高能量需求時,循環中乳酸是除大腦外所有組織TCA循環的主要碳源。因此,細胞糖酵解代謝增加可以快速提供細胞活化所需的能量和生物大分子物質。與腫瘤細胞類似,糖酵解增加是免疫細胞快速活化的標志性代謝改變[13]。M1型巨噬細胞、活化的樹突狀細胞和Th17細胞等被炎癥因子激活,細胞糖酵解代謝增加,能夠快速產生ATP和生物大分子物質來執行其特定的效應功能,如巨噬細胞的吞噬作用、炎性細胞因子產生、樹突狀細胞的抗原呈遞和Th17細胞的效應細胞因子IL-17產生[14]。
2 巨噬細胞糖酵解代謝
細胞代謝在巨噬細胞活化過程中發揮著決定性作用。M1和M2型巨噬細胞利用不同的代謝方式為其效應功能提供能量。M1型巨噬細胞利用糖酵解代謝為劇烈、短暫的殺菌或促炎反應迅速提供能量;而M2型巨噬細胞利用線粒體氧化磷酸化和脂肪酸氧化(fatty acid oxidation,FAO)長時間、持續地生成ATP,為組織修復提供助力[15]。這些代謝差異最初被認為反映細胞的底物利用,但現在研究認為細胞能量代謝轉變可以直接改變巨噬細胞的極化狀態和炎癥反應。巨噬細胞代謝轉向糖酵解,促進炎性M1型巨噬細胞表型[16]。2-脫氧-D-葡萄糖(2-deoxy-D-glucose,2DG)是一種葡萄糖類似物,能夠競爭性地抑制糖酵解,研究證實2DG通過阻斷M1型巨噬細胞糖酵解,抑制其炎癥表型包括吞噬功能、炎性細胞因子分泌及活性氧簇(reactive oxygen species,ROS)產生[16-17]。而另有研究證實,乙莫克舍(etomoxir)是一種FAO代謝關鍵酶肉毒堿棕櫚酰基轉移酶1α(carni?tine palmitoyl transferase-1α,CPT-1α)抑制劑,可以降低M2型巨噬細胞的線粒體耗氧率和儲備呼吸能力,從而抑制M2型巨噬細胞的線粒體氧化磷酸化,并減少其表面標志物CD301、CD206和RELMα的表達[18]。過表達巨噬細胞過氧化物酶體增殖物激活受體γ輔激活因子1β可驅動線粒體氧化磷酸化,促進M2型巨噬細胞激活并抑制炎癥因子產生[19]。因此,通過干預代謝方式調控巨噬細胞的極化狀態和炎癥反應有利于炎癥性疾病的治療。
目前調控炎性巨噬細胞糖酵解上調的機制已基本明確。Rodríguez-Prados等[15]使用基于葡萄糖示蹤物的代謝組學方法證實,多種Toll樣受體(Toll-like receptors,TLRs)途徑激活均可增加小鼠腹腔原代巨噬細胞糖酵解代謝。以被廣泛研究的TLR4配體LPS和干擾素γ(interferon-γ,IFN-γ)刺激的M1型巨噬細胞為例,調控炎性巨噬細胞糖酵解代謝的機制主要包括3種途徑:(1)直接改變糖酵解蛋白表達水平:LPS促進6-磷酸果糖-2-激酶/果糖-2,6-二磷酸酶(6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase,PFK2)亞型表達發生變化,從肝臟形式(liver form,LPFK2)轉為更活躍的普遍存在形式(ubiquitous form,u-PFK2),而u-PFK2催化6-磷酸果糖生成2,6-二磷酸果糖,后者的濃度增加可激活糖酵解關鍵酶6-磷酸果糖激酶1(phosphofructokinase 1,PFK1),增加糖酵解通量[15]。同時,LPS快速下調巨噬細胞碳水化合物激酶樣蛋白(carbohydrate kinase-like protein,CARKL),從而增加PPP通量,維持M1型巨噬細胞合成代謝[20]。(2)缺氧誘導因子(hypoxia-inducible factor,HIF)依賴性途徑:Blouin等[21]證明LPS刺激巨噬細胞HIF-1α蛋白穩定表達,并形成功能性異二聚體復合物,與糖酵解相關基因和炎癥基因的缺氧反應元件結合,激活炎性M1型巨噬細胞表型。同時,NF-κB、AKT/mTOR等炎癥通路同樣可以調節HIF-1α表達,促進糖酵解代謝[22-24]。另外,M1型巨噬細胞TCA循環中2個環節受損,導致琥珀酸和檸檬酸累積,過量的琥珀酸抑制脯氨酸羥化酶的活性,從而維持HIF-1α穩定[25]。而檸檬酸轉運到細胞質生成NADPH,促進炎癥介質ROS生成,ROS具有穩定HIF-1α表達的作用,HIF-1α增加進一步激活糖酵解基因的轉錄,從而維持M1型巨噬細胞糖酵解代謝[26]。此外,存在LPS刺激的情況下,缺氧激活HIF-1α通路更顯著增加巨噬細胞糖酵解通量[6]。(3)非HIF依賴性途徑:LPS激活巨噬細胞信號級聯反應募集腫瘤壞死因子受體相關因子6(tumor necrosis factor receptor-associ?ated factor 6,TRAF6)和TANK結合激酶1(TANK-binding kinase-1,TBK-1),TBK-1磷酸化信號轉導及轉錄激活因子3(signal transducer and activator of transcription 3,STAT3)的第727位絲氨酸殘基,促進M1型巨噬細胞糖酵解增加和炎性細胞因子的產生[27]。Hedl等[28]證實,LPS激活干擾素調節因子5(interferon regulatory factor 5,IRF5)-AKT2信號通路,M1型巨噬細胞糖酵解增加,并促進M1型巨噬細胞極化。
3 單核-巨噬細胞糖酵解增加促進AS進展
在AS早期病變時,骨髓造血細胞的糖酵解促進髓系細胞生成,導致循環中單核細胞在斑塊的積累增加,間接促進斑塊病變生長[8,29]。而單核細胞的糖酵解增加同樣促進AS的進展。與健康個體的循環單核細胞相比,AS患者的循環中單核細胞具有更高的糖酵解通量,其中單核細胞糖酵解增加程度與線粒體活性氧生成、氧化應激和炎癥信號激活程度成正比[6]。在進展性病變中,斑塊內巨噬細胞在氧化脂質、氧化應激和缺氧等刺激下,穩定表達HIF-1α,并且和葡萄糖轉運蛋白1(glucose transporter 1,GLUT1)、己糖激酶2(hexokinase 2,HK2)和丙酮酸脫 氫 酶 激 酶1(pyruvate dehydrogenase kinase 1,PDK1)的表達同步,表明斑塊巨噬細胞糖酵解代謝增強。此外,在斑塊內富含巨噬細胞區域IL-1β同樣高表達[30-31],說明糖酵解增加參與AS斑塊炎癥進展。另一項動物研究中,骨髓特異性敲除Hif-1α低密度脂蛋白受體敲除(low-density lipoprotein receptor knockout,Ldlr-/-)小鼠的AS斑塊形成被抑制,進一步證實了HIF-1α及其介導的糖酵解在AS進展中的促進作用[32]。
然而,另有研究指出適度的糖酵解過程可以維持巨噬細胞吞噬功能,過度抑制糖酵解反而加劇AS進展。Morioka等[33]的研究證實,Slc2a1基因(編碼GLUT1的基因)敲除小鼠的骨髓源性巨噬細胞表現出胞吞能力下降;將髓系Slc2a1缺陷小鼠的骨髓移植到Ldlr-/-小鼠骨髓內,高脂喂養12周后,小鼠主動脈根部斑塊壞死核心區面積顯著增加,壞死核心區內凋亡細胞的數量也顯著增加,表明斑塊內Slc2a1缺失巨噬細胞有效吞噬功能下降。因此,適度的糖酵解也是調節巨噬細胞有效吞噬的關鍵,可以防止形成斑塊壞死核心區,避免易損斑塊破裂。
4 抑制丙酮酸激酶M 2(pyruvate kinase M 2,PKM 2)通過介導糖酵解減輕AS進展
PKM2是糖酵解最后一個關鍵限速酶,在腫瘤細胞中特異性地高表達。PKM2主要以2種構型存在:四聚體形式(非活躍狀態)和二聚體形式(活躍狀態)。四聚體PKM2具有糖酵解酶活性,催化磷酸烯醇式丙酮酸生成丙酮酸。二聚體PKM2具有磷酸激酶活性,通過轉移進入細胞核與具有調控活性的蛋白相互作用從而促進靶基因的轉錄。此外,二聚體PKM2可以被分泌至細胞外,血漿和糞便中的PKM2可作為大腸癌、乳腺癌等的診斷標志物[34]。
近年,PKM2被證實在炎性巨噬細胞中亦高表達,同時介導免疫相關功能。PKM2不僅是受HIF-1α調控的靶基因,它還直接與HIF-1α相互作用并促進HIF-1α調控基因的轉錄。Palsson-McDermott等[35]發現,LPS誘導巨噬細胞二聚體PKM2轉移入細胞核,作為HIF-1α的結合蛋白,促進糖酵解基因和炎癥基因IL-1β轉錄(圖1);選擇性PKM2小分子激活劑TEPP-46(ML265)可以恢復PKM2酶活性,促使PKM2四聚體化,并阻止二聚體PKM2核轉位,還可以抑制LPS誘導的巨噬細胞糖酵解和IL-1β表達,從而促進M1型巨噬細胞向M2型轉化。同時,PKM2促進巨噬細胞NLRP3(NLR family pyrin domain con?taining 3)炎癥小體和AIM2(absent in melanoma 2)炎癥小體的激活;PKM2依賴的糖酵解可促進RNA依賴性蛋白激酶(RNA-dependent protein kinase,PKR)的磷酸化,從而誘導巨噬細胞NLRP3炎癥小體激活,分泌IL-1β、IL-18和高遷移率族蛋白B1[36-37]。
PKM2最初通過增加腫瘤細胞Warburg效應促進腫瘤生長。Shirai等[7]證實,PKM2同樣參與AS的發生,冠心病患者循環單核細胞體外分化的巨噬細胞表現出二聚體PKM2表達增加、糖酵解通量增加和ROS生成上調,二聚體PKM2轉位到細胞核,磷酸化STAT3,導致下游炎癥因子IL-1β和IL-6表達增加。Kumar等[38]證實,oxLDL誘導巨噬細胞源性泡沫細胞中PKM2第105位酪氨酸殘基磷酸化,并促進PKM2轉位入核,增加HIF-1α靶基因Ldha、Slc2a1和Il-1β的轉錄及IL-1β的分泌,從而促進糖酵解代謝和炎癥反應。紫草素(shikonin,SKN)是PKM2的有效抑制劑,通過抑制巨噬細胞糖酵解代謝發揮抗炎作用[36,39]。SKN可以減緩高同型半胱氨酸血癥誘導的載脂蛋白E敲除(apolipoprotein E knockout,ApoE-/-)小鼠AS的形成,降低了主動脈斑塊內炎癥因子TNF-α、IL-1β和ICAM-1的表達。這種治療作用主要由于SKN對PKM2的抑制,從而減弱了細胞活化所需的糖酵解代謝[40]。因此,糖酵解限速酶PKM2是減輕巨噬細胞炎癥、預防AS的新靶點。
5 靶向降低巨噬細胞糖酵解代謝水平可減輕AS炎癥
降脂治療雖然有效降低了心血管疾病的患病風險,但是患者仍存在殘余風險,部分原因是低度慢性炎癥未減輕[41]。CANTOS試驗顯示,用canakinumab靶向IL-1β可降低事件復發的風險,表明抗炎策略在AS治療中具有很大的應用前景[42]。直接的全身抗炎治療有影響機體免疫功能及增加腫瘤風險的可能性,而干預巨噬細胞代謝的方式提高了抗炎治療的靶向性。
二甲雙胍作為2型糖尿病的一線治療藥物,通過調節胰島素靶細胞線粒體功能和能量傳感器AMP活化蛋白激酶(AMP-activated protein kinase,AMPK)活性來改善葡萄糖和脂肪酸代謝[43]。He等[44]的研究表明,二甲雙胍可減輕oxLDL誘導的脂質氧化,增強GLUT1介導的葡萄糖轉運,并且增加線粒體氧化代謝,從而誘導炎性巨噬細胞向抗炎表型轉變。單核細胞受到炎性刺激如oxLDL,通過代謝和表觀遺傳重編程誘導持久的促炎癥、致AS表型稱為訓練免疫[45]。Keating等[46]的研究評估了糖酵解基因變異對單核細胞訓練免疫能力的影響,結果顯示糖酵解酶磷酸果糖激酶2/果糖-2,6-二磷酸酶3(phosphofructo?kinase-2/fructose-2,6-bisphosphatase 3,PFKFB3)和血小板型磷酸果糖激酶(the platelet isoform of phos?phofructokinase,PFKP)的突變可以降低oxLDL誘導的訓練免疫能力;而體內給予二甲雙胍使oxLDL誘導人外周血單核細胞訓練免疫的能力減弱。上述研究提示二甲雙胍作為AS抗炎治療藥物的可能性。但是二甲雙胍作為降糖藥物可導致患者乳酸中毒,并且目前臨床只適用于高血糖患者的AS防治。
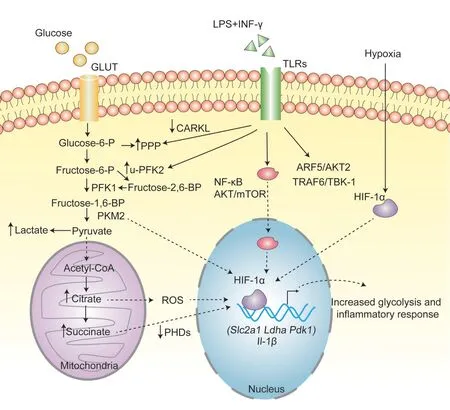
Figure 1.Schematic diagram of the mechanism of glycolysis regulation in inflammatory M1 macrophages.Glucose uptake increases in M1 macrophages stimulated by LPSand IFNγ.LPSincreases glycolytic flux through down-regulating CARKL protein ex?pression and up-regulating u-PFK2 expression.LPSalso impairs the TCA cycle,and causes the accumulation of citrate and succinate.Citrate is transported to the cytoplasm to generate NADPH,which promoting ROSgeneration.ROSand succi?nate stablize the expression of HIF-1α,thereby increasing the transcription of target genes related to glycolysis and IL-1β.Simultaneously,inflammatory pathways such as NF-κB and AKT/mTOR regulate the expression of HIF-1αto promote gly?colytic metabolism.In addition,with the stimulation of LPS,hypoxia significantly enhances glycolytic metabolism by acti?vating the HIF-1αpathway.Finally,LPSactivates signaling cascade to recruit TRAF6-TBK-1 protein and activates the IRF5-AKT2 signaling through a non-HIF-1α-dependent pathway,leading to macrophage glycolysis enhancement and fol?lowing activation of M1 macrophage inflammation.圖1 炎性M 1巨噬細胞糖酵解代謝調節機制的示意圖
微小RNA(microRNA,miRNA,miR)參與調控巨噬細胞極化被廣泛報道[47]。Ouimet等[48]證實,miR-33通過改變巨噬細胞代謝方式調節細胞炎癥表型;抑制巨噬細胞miR-33表達可以激活AMPK通路,上調FAO,并減弱糖酵解代謝,從而誘導巨噬細胞轉化為M2表型;體內抗miR-33治療可以減少小鼠主動脈斑塊面積;通過激光捕獲顯微切割分離Ldlr-/-小鼠斑塊病變巨噬細胞,與對照組相比,抗miR-33治療后小鼠斑塊內M2型巨噬細胞標志基因(Arg1和Mrc1)富集,而M1型巨噬細胞標志基因(Ifnb1、Il1a和Nox1)表達減少。抗miR-33治療雖然可以誘導巨噬細胞轉化為M2表型,但不能排除對體內其他類型細胞的影響,且目前尚缺乏臨床試驗證明其安全性及有效性。
6 總結與展望
巨噬細胞的表型轉化和炎癥反應與細胞糖酵解代謝的關系緊密交織,炎性巨噬細胞糖酵解的增加促進AS斑塊的起始、進展及轉歸。研究顯示,二甲雙胍和miRNA抑制劑等可以降低巨噬細胞糖酵解速率,增加線粒體氧化代謝,抑制炎癥反應,從而減輕AS,提示降低炎性巨噬細胞糖酵解水平可能成為AS的治療策略。但是,目前從抑制巨噬細胞糖酵解出發,開展AS防治研究仍面臨眾多挑戰,例如在體斑塊內巨噬細胞代謝表型鑒定技術仍不完善、斑塊內巨噬細胞糖酵解代謝相關分子靶點仍有待探索等。因此,深入探究炎性巨噬細胞糖酵解相關分子靶點并開發在體代謝表型檢測技術,有望為研究者從抑制AS中巨噬細胞糖酵解代謝出發穩定和逆轉AS提供理論基礎和有效技術手段。
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
東方少年·布老虎畫刊(2023年8期)2023-08-01 15:45:12
科學大眾(2021年6期)2021-07-20 07:42:44
科學(2020年3期)2020-11-26 08:18:30
學苑創造·A版(2020年9期)2020-10-13 09:41:02
娃娃樂園·綜合智能(2019年3期)2019-04-03 09:17:36
中成藥(2018年2期)2018-05-09 07:19:34
小學生學習指導(低年級)(2017年10期)2017-10-10 01:00:05
湖北師范大學學報(自然科學版)(2015年2期)2016-01-10 08:41:55
云南中醫學院學報(2014年3期)2014-07-31 18:57:34
