絡合物法制備鎳-氮共摻雜炭基二氧化碳電催化劑
2021-09-09 07:10:36常若鵬郝廣平
無機鹽工業 2021年9期
常若鵬,胡 旭,賀 雷,郝廣平
(大連理工大學化工學院,精細化工國家重點實驗室,遼寧省低碳資源高值化利用重點實驗室,遼寧 大連 116024)
近年來,大氣中二氧化碳(CO2)濃度的增加使得地球溫室效應不斷加劇,隨之引發的冰川融化、海平面上升等問題正嚴重威脅著人類未來的生存空間。因此,如何有效降低大氣中CO2的濃度已成為人類社會可持續發展過程中必須要面對的問題[1-2]。二氧化碳電催化還原反應(CO2RR)可以在溫和條件下將CO2轉化為一氧化碳、甲酸等化學品,同時實現了碳資源的循環與清潔能源的儲存,該課題引起了人們的關注[3-4]。而設計出高選擇性、高活性的電催化劑是推動CO2RR實現規模化應用的關鍵。
利用炭材料自身來源廣泛、導電性好、結構穩定等優勢,研究者們通過形貌調控以及雜原子摻雜等方式對炭材料進行功能化處理,制備了一系列摻雜型炭基電催化劑。其中,過渡金屬-氮共摻雜的炭基催化劑因良好的催化能力及低廉的價格得到了大量報道。按照制備方法的不同可以分為:1)混合物直接炭化法[5-7];2)金屬摻雜g-C3N4作前驅體炭化法[8-9];3)高溫熱解金屬-有機框架材料(MOFs)法[10-12];4)氮摻雜炭載體負載金屬原子法[13-15];5)炭載體錨定金屬絡合物法[16-17]。其中,炭載體錨定金屬絡合物法是利用過渡金屬與含氮配體形成的絡合物與炭載體進行復合,該方法可以在溫和條件下將金屬絡合物錨定在炭載體框架中,一步獲得過渡金屬-氮共摻雜的炭基催化劑。目前,卟啉、酞菁及鄰二氮菲等含氮大分子基團通常會被選擇作為配體與過渡金屬鹽溶液進行配位反應形成穩定的配位溶液與炭載體進行直接負載,獲得了較好的催化效果。然而,卟啉、酞菁及鄰二氮菲等物質價格昂貴,高負載下的金屬絡合物容易形成團聚,導致催化劑性能降低。因此,筆者對傳統的炭載體錨定金屬絡合物法進行改進,將廉價的乙二胺與硝酸鎳形成的小分子絡合物溶液作為鎳源及氮源,分別以親水氧化石墨烯及疏水炭黑作為載體,對絡合物負載的炭載體進行炭化、酸洗處理,得到鎳-氮共摻雜催化劑。通過X射線衍射、透射電子顯微鏡等手段表征不同處理階段鎳物種的存在形式,結合同一載體催化劑在炭化前后及酸洗前后催化性能上的變化,得出高度分散的鎳物種對二氧化碳電催化反應具有促進效果。對比不同炭載體酸洗后樣品的催化性能,發現使用疏水性炭載體有助于提高絡合物在炭載體骨架間的分散程度,進而提高催化劑的催化活性。
1 實驗部分
1.1 材料制備
過渡金屬絡合物溶液的制備:配制10 mL各類過渡金屬鹽溶液(0.1 mol/L),按照1∶3物質的量比分別向其中加入對應體積的哌嗪溶液及乙二胺溶液,靜置24 h;按照1∶1、1∶3、1∶5、1∶7物質的量比分別向10 mL 0.1 mol/L的Ni(NO3)2溶液中加入對應體積的哌嗪溶液及乙二胺溶液,靜置24 h。記錄各絡合物溶液的狀態。
親水性鎳-氮共摻雜催化劑的制備:按照1∶3物質的量比取10 mL 0.1 mol/L的Ni(NO3)2溶液與200 μL乙二胺(EN)混合,得到紫色的三乙二胺合鎳絡合物溶液,將其與20 mL氧化石墨烯(GO,3 mg/mL)分散液混合,在180℃水熱處理24 h,得到的固相產物命名為Ni-EN-GO;將Ni-EN-GO在900℃炭化處理2 h,得到的炭化產物命名為Ni NPs@Ni-NG;將Ni NPs@Ni-NG經過24 h鹽酸酸洗處理(4 mol/L),得到以石墨烯為炭載體的親水性鎳-氮共摻雜催化劑,命名為Ni-NG。
疏水性鎳-氮共摻雜催化劑的制備:采用等體積浸漬法,將0.3 mL三乙二胺合鎳絡合物溶液負載于100 mg科琴黑載體中,干燥后的樣品命名為Ni-EN-KB;將Ni-EN-KB在900℃炭化處理2 h,得到的炭化產物命名為Ni NPs@Ni-NKB;將Ni NPs@Ni-NKB經過24 h鹽酸酸洗處理(4 mol/L),得到以科琴炭黑為炭載體的鎳-氮共摻雜催化劑,命名為Ni-NKB。采用同樣的方法,按照20%的理論鎳含量(質量分數)制備疏水性鎳-氮共摻雜催化劑,將酸洗處理前后的樣品分別命名為Ni NPs@Ni-NKB-2、Ni-NKB-2。
工作電極的制備:稱取9 mg催化劑,分散在由665 μL去離子水、285 μL無水乙醇以及50 μL Nafion組成的溶液中(質量分數為5%),分散液超聲處理1 h得到催化劑勻漿;取10 μL催化劑勻漿,分兩次滴加在玻碳電極表面(玻碳電極直徑為3 mm),使用紅外干燥燈烘干,得到負載催化劑的玻碳電極。取300 μL催化劑勻漿,使用移液槍分6次滴加在圓形碳紙兩面(圓形碳紙直徑為1.2 cm),在室溫干燥4~5 h,然后轉移至80℃真空干燥箱中干燥過夜,得到催化劑負載的碳紙。
1.2 測試表征
采用D/Max 2400 X射線粉末衍射儀(XRD)對樣品進行物相分析;采用S-4800型掃描電子顯微鏡(SEM)及Tecnai F30型透射電子顯微鏡(TEM)觀察樣品的形貌;采用Vairo EL cube型元素分析儀對樣品體相元素含量進行檢測,樣品中金屬元素含量由Nex ION 300D電感耦合等離子體-原子發射光譜儀(ICP-AES)進行測試;采用Elexsys 580型電子順磁共振儀對樣品進行電子順磁共振(EPR)測試;采用HARKE-SPCA型接觸角測試儀對樣品接觸角進行測試;材料的電催化性能測試由Ivium-Stat工作站完成,氣相產物檢測由GC7900型氣相色譜儀完成。
2 實驗結果與討論
首先,對不同種類的過渡金屬鹽與配體的配位反應規律進行探究,希望通過含氮配體與金屬原子形成穩定的絡合物溶液,在溫和條件下將金屬絡合物一步負載于炭載體上,進而獲得過渡金屬-氮共摻雜的炭基催化劑。選擇哌嗪(C4H10N2)與乙二胺(C2H8N2)作為配體,按照過渡金屬與配體物質的量比為1∶3將配體分別加入至鎳、銅、鐵、鈷、錳5種過渡金屬鹽溶液中,不同過渡金屬鹽與配體間的配位反應見表1。

表1 過渡金屬鹽與配體的配位反應規律Table 1 Regulation of coordination reactions between transition metals and ligands
將配體加入金屬鹽溶液后,溶液顏色迅速發生變化,說明各過渡金屬與配體間發生了明顯的配位反應。由于各絡合物在水中的溶解度存在差異,經過長時間靜止,部分絡合物以沉淀形式析出,溶液出現明顯的分層現象。值得注意的是,乙二胺與硝酸鎳的配位體系較為穩定,經過長時間靜止仍為均勻溶液,有利于在水溶液體系下進行絡合物的負載。此外,按照硝酸鎳與乙二胺物質的量比為1∶1、1∶5、1∶7進行配位實驗,同樣可以獲得穩定的絡合物溶液。因此,使用乙二胺作為配體、硝酸鎳作為鎳源獲得的鎳配合物作為鎳源與氮源,與炭載體進行復合,即可得到鎳-氮共摻雜炭基催化劑前驅體。
為確定硝酸鎳與乙二胺的配比,按照硝酸鎳與乙二胺物質的量比分別為0∶3、1∶3、1∶7制備前驅體,對3組前驅體進行電子順磁共振(EPR)測試,結果見圖1。由圖1看出,在物質的量比為0∶3及1∶7時,在磁場強度為267 100 A/m附近出現的峰對應未發生配位作用的氮原子上的未成對電子,隨著乙二胺用量增加未配位的氮原子數增多,對應峰強度明顯增加。在物質的量比為1∶3時,相同位置處的峰消失,說明在該配比下乙二胺中所有氮原子均與鎳原子發生配位作用,形成三乙二胺合鎳配合物,這個結果與有關文獻報道的結果相同[18]。因此,選擇硝酸鎳與乙二胺物質的量比為1∶3。
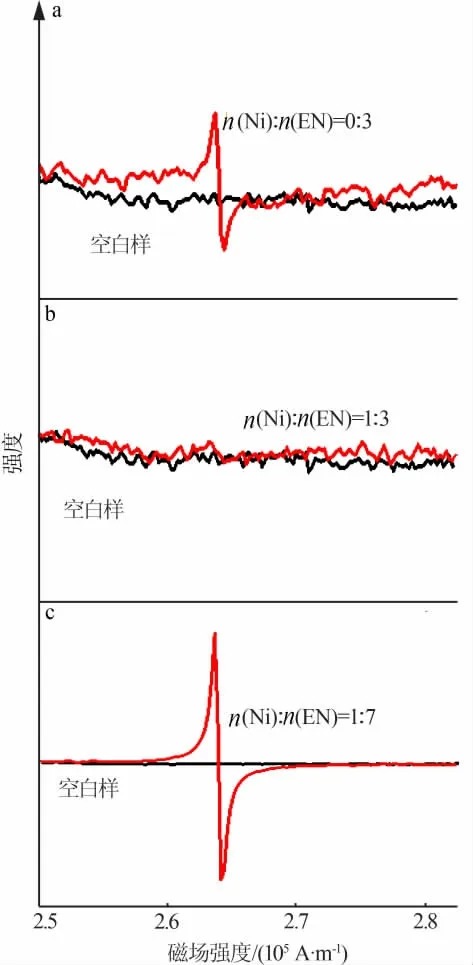
圖1 不同n(Ni)∶n(EN)所得前驅體EPR測試圖Fig.1 EPR spectra of precursor with varied n(Ni):n(N)
將硝酸鎳與乙二胺物質的量比為1∶3形成的三乙二胺合鎳絡合物作為鎳源與氮源,分別以親水氧化石墨烯及疏水炭黑作為載體制備催化劑,兩種樣品XRD譜圖見圖2。圖2表明,經過900℃炭化處理,樣品Ni NPs@Ni-NG位于2θ為10.8°的氧化石墨烯特征峰消失,在2θ為26°處出現了對應石墨烯(002)晶面的特征衍射峰,同時在2θ為44.4、51.8°處出現了對應鎳金屬(111)(002)晶面的特征衍射峰。說明經過180℃水熱處理及900℃炭化處理,氧化石墨烯被還原成為石墨烯,樣品中部分鎳物種發生團聚形成鎳金屬顆粒。酸洗處理后,鎳金屬顆粒已基本被除去,對應的特征衍射峰消失。經過900℃炭化處理,樣品Ni NPs@Ni-NKB中同樣出現了對應鎳金屬的特征衍射峰;酸洗處理后團聚態的鎳被除去,樣品Ni-NKB中僅出現了位于2θ為26、44°處的峰,分別對應炭材料的(001)(101)晶面。
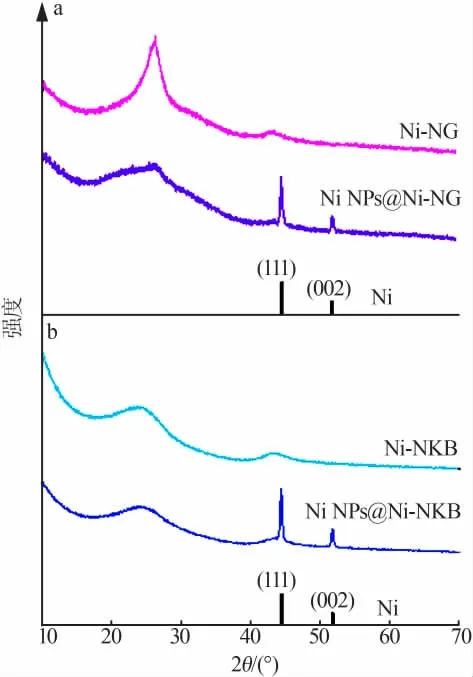
圖2 GO體系樣品(a)和KB體系樣品(b)XRD譜圖Fig.2 XRD patterns of the samples of GO system(a)and KB system(b)
采用SEM和TEM對Ni-NG樣品和Ni-NKB樣品進行形貌表征,結果見圖3。由圖3a~d可見,Ni-NG樣品的SEM照片顯示材料的主體為堆疊的石墨烯片層結構,與TEM照片中出現的石墨烯特有的褶皺結構相對應。同時,無論在SEM照片或TEM照片中均未發現明顯的鎳金屬顆粒,說明酸洗處理后Ni-NG樣品中鎳物種應以高度分散的狀態存在。由圖3e~h可見,Ni-NKB樣品的SEM照片中可以觀察到尺寸分布均勻的納米炭顆粒,顆粒之間堆積緊密,提高了材料的導電性;TEM照片中同樣未發現明顯的鎳金屬顆粒,說明Ni-NKB樣品中鎳物種也應以高度分散的形式存在,與XRD測試結果相符。根據文獻報道,在炭載體中高度分散的金屬原子具有較好的CO2電催化活性,有利于提高產物的法拉第效率,抑制析氫反應的發生[19]。

圖3 Ni-NG樣品和Ni-NKB樣品電鏡照片Fig.3 SEM and TEM images of Ni-NG and Ni-NKB
為進一步確定樣品中的元素含量,使用元素分析及ICP-AES對樣品Ni-NG進行表征。結果表明樣品Ni-NG的體相元素組成(原子分數):Ni,0.45%;N,4.92%;C,83.76%;H,5.80%。可以看出,將三乙二胺合鎳絡合物負載于氧化石墨烯載體上,在炭骨架中引入鎳、氮元素,經過進一步炭化、酸洗處理,獲得了鎳-氮共摻雜催化劑。
為比較樣品的親、疏水性差異,測試了Ni-NG樣品和Ni-NKB樣品的動態接觸角,結果見圖4。由圖4看出,Ni-NG與水之間形成的接觸角接近43.5°,具有較為明顯的親水特性,這是由于其炭載體氧化石墨烯在炭化處理過程中并未被完全還原造成的(O原子分數為5.16%);Ni-NKB采用疏水炭黑作載體,其接觸角接近67.0°,顯示出了一定的疏水特性。相比之下,親水性炭載體容易與絡合物間形成較強的作用力,使絡合物在炭載體中的負載較為集中,在炭化過程中更容易出現鎳物種的團聚;疏水性炭載體與絡合物溶液之間作用力相對較弱,有利于絡合物在炭載體中的分散,炭化過程中鎳物種不易團聚,減少了炭化酸洗處理后鎳物種的損失。
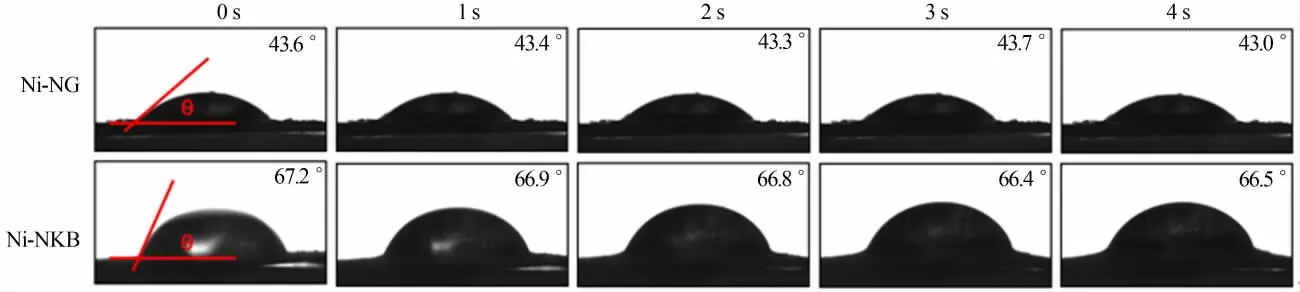
圖4 Ni-NG樣品與Ni-NKB樣品的接觸角測試圖Fig.4 Images of contact angle measurements of Ni-NG and Ni-NKB
為進一步證明親、疏水性不同的炭載體催化劑在炭化過程中鎳物種團聚程度的不同,對Ni NPs@Ni-NKB-2樣品和Ni-NKB-2樣品進行了XRD表征,結果見圖5。由圖5看出,Ni NPs@Ni-NKB-2樣品(理論鎳質量分數為20%)在酸洗處理后仍可以檢測到鎳金屬的特征衍射峰,而具有相似理論鎳含量(理論鎳質量分數為20.87%)的Ni NPs@Ni-NG樣品在酸洗處理后對應鎳的特征衍射峰消失(圖2)。兩組樣品間的差異表明,在炭化過程中不同催化劑中鎳物種的團聚情況不同。在高負載量條件下,由于氧化石墨烯的親水性作用,鎳物種與氧化石墨烯的極性位點相互作用較強,熱解過程中高密度鎳物種容易面內遷移,進而團聚形成較大尺寸的鎳金屬顆粒。由于其二維開放結構,酸洗處理后鎳納米顆粒基本完全除去;而以疏水多孔炭黑作為載體的催化劑中,鎳物種在多孔骨架上分散程度較好,由于炭質孔壁的保護作用,酸洗處理并不能完全將小尺寸的鎳物種除去。
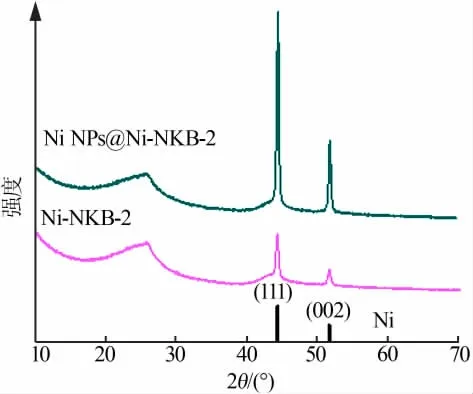
圖5 NiNPs@Ni-NKB-2樣品與Ni-NKB-2樣品XRD譜圖Fig.5 XRD patterns of the Ni NPs@Ni-NKB-2 and Ni-NKB-2
為測試各階段樣品的催化性能,使用氣密性良好的H型電解池對各階段樣品的電化學性能進行測試。首先,在-1.0~-0.2 V(vs.RHE)電位區間進行線性伏安掃描(LSV)測試,結果見圖6a、b。由圖6a看出,在GO體系中水熱處理后的樣品Ni-EN-GO在CO2氣氛下電催化反應的電流密度基本與在Ar氣氛下的電流密度相同,說明樣品沒有CO2還原活性,電解反應的電流密度全部由析氫反應提供;而經過900℃處理的樣品Ni NPs@Ni-NG在CO2氣氛下的電流密度明顯提高,說明樣品中形成了具有CO2催化還原活性的活性中心(鎳-氮-碳結構),同時樣品具有較大的析氫反應電流密度,可能是因為樣品中鎳金屬顆粒的存在;通過酸洗處理除去鎳金屬顆粒后,樣品Ni-NG在CO2氣氛下的電流密度基本不變,析氫反應電流密度降低。同樣由圖6b看出,在KB體系中經過炭化處理各階段樣品都出現了CO2催化還原活性,與Ni NPs@Ni-NKB相比酸洗后Ni-NKB樣品的反應電流密度明顯降低,可能是因為Ni NPs@Ni-NKB樣品中含有大量的鎳顆粒,因此反應電流密度主要由析氫反應提供,在酸洗處理后鎳顆粒被除去,析氫反應所提供的電流密度迅速減小,所以無論是在Ar氣氛下的電流密度還是在CO2氣氛下的電流密度都會出現明顯降低。
由于反應產物主要是一氧化碳(CO)和氫氣(H2),因此為測試各樣品電催化反應中CO產物的法拉第效率,在不同的電位下對材料進行恒電位電解處理。在同一電位下電解1 h后檢測產物中CO與H2的含量,進而計算在該電位下不同產物的法拉第效率,結果見圖6c、d。900℃炭化處理的Ni NPs@Ni-NG樣品和Ni NPs@Ni-NKB樣品的CO法拉第效率最大值僅為70%和60%,這是因為樣品中團聚態的鎳物種有利于析氫反應的進行,促進了產物氫氣的生成,使得樣品CO法拉第效率降低。Ni-NG樣品在-0.8 V(vs.RHE)的CO法拉第效率達到最高值90%,在-0.9~-0.7 V(vs.RHE)的CO法拉第效率均處于80%以上。Ni-NKB樣品在-0.8 V(vs.RHE)的CO法拉第效率同樣達到最高值92%,說明兩樣品的活性中心可能是相似的。對比除去鎳顆粒后樣品的CO選擇性,在-0.7~-0.6 V(vs.RHE),Ni-NG的CO法拉第效率高于Ni-NKB;在-1.2~-0.9 V(vs.RHE),Ni-NKB的CO法拉第效率高于Ni-NG。兩樣品法拉第效率上的差異可能是炭載體的親、疏水特性影響了析氫反應發生的電位,與Ni-NG相比以疏水炭黑作炭載體的Ni-NKB發生析氫反應需要克服更高的能壘,因此反應電位向更高的電位移動。
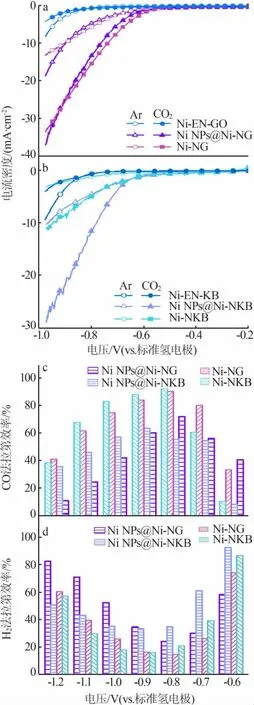
圖6 各階段樣品電催化CO2還原性能測試圖Fig.6 Electrocatalytic CO2RR performance of the prepared samples
在電催化反應中,電流密度一般由材料中活性位的密度決定,是評價催化劑催化性能的一項重要參數。根據恒電位測試結果及測試用電極片負載的催化劑質量,作出不同電位下各樣品的分電流密度圖,結果見圖7。由圖7a可見,酸洗后的Ni-NG、Ni-NKB樣品與未酸洗的Ni NPs@Ni-NG、Ni NPs@Ni-NKB樣品在CO分電流密度上存在明顯差異,基本趨勢均為酸洗樣品CO分電流密度大于未酸洗樣品。在-1.0 V(vs.RHE),Ni-NKB樣品的CO分電流密度達到最高,為7.2 mA/g。樣品Ni-NG具有相似的規律,在-1.1 V(vs.RHE),其CO分電流密度達到最高,為6.4 mA/g。兩樣品在-1.2 V(vs.RHE)CO分電流密度下降、H2分電流密度增加,這是由于高電位下析氫反應明顯增強造成的。值得注意的是,Ni NPs@Ni-NKB樣品隨著電位的提高CO分電流密度不斷增加,并未出現類似Ni NPs@Ni-NG樣品的CO分電流密度變化趨勢。產生這種差異的原因可能是因為疏水性炭載體使得析氫反應的優勢電位向高電位移動,因此即使在-1.2~-1.1 V(vs.RHE),樣品的CO分電流密度仍可以出現小幅度提高。而以親水氧化石墨烯作炭載體的Ni NPs@Ni-NG樣品,隨著電位的不斷提高析氫反應不斷增強,CO分電流密度會迅速下降。從各樣品的H2分電流密度圖(圖7b)可以看出,其基本趨勢為未酸洗樣品H2分電流密度大于酸洗樣品,這進一步說明了高度分散態的鎳物種有利于CO2電催化還原反應的發生,而團聚態的鎳物種有利于析氫反應的進行。兩體系樣品的CO分電流密度也同樣存在差異,相較于Ni-NG在相同電位下Ni-NKB樣品的CO分電流密度更大,具有更好的催化活性。這種差異可能是因為在水溶液體系中,三乙二胺合鎳絡合物與親水氧化石墨烯表面的含氧官能團間具有較強的共價作用,炭化過程中鎳物種更容易發生團聚。而疏水炭黑與絡合物溶液間的相互關系較弱,在負載過程中有利于鎳物種的分散,避免了酸洗過程中鎳物種大量損失,催化劑的催化活性較高。
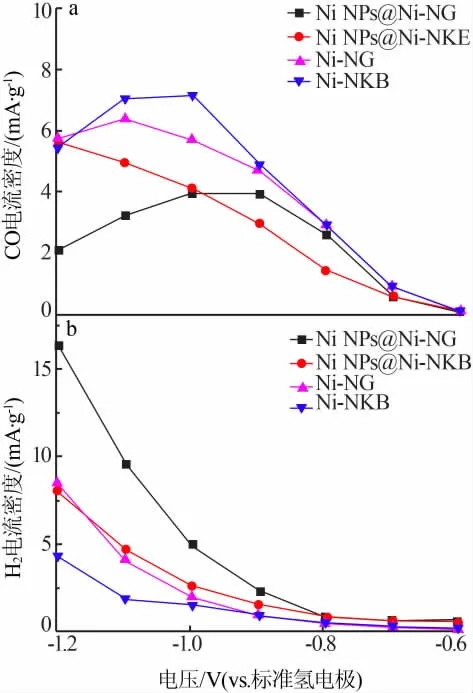
圖7 不同電位下CO與H2的分電流密度Fig.7 Current density of CO and H2 at varied potentials
3 結論
選擇乙二胺作為小分子含氮配體與硝酸鎳反應得到三乙二胺合鎳絡合物溶液,將其分別負載于疏水炭黑與親水氧化石墨烯炭載體中,獲得了親、疏水具有明顯差異的兩類鎳-氮共摻雜催化劑。在同一炭載體條件下,酸洗處理前后的樣品在電催化性能上出現明顯提升,說明高度分散的鎳物種對于CO2電催化還原為CO具有促進作用。此外,親、疏水不同的炭載體與金屬絡合物間的結合方式不同,親水氧化石墨烯表面的含氧官能團與過渡金屬絡合物間會形成較強的共價作用,疏水炭黑則與絡合物溶液間的相互作用較弱,相比之下更有利于絡合物溶液分散在炭骨架中,減少在炭化處理中出現鎳物種因為團聚形成鎳金屬顆粒而造成活性物質的損失,在相同電位下獲得更高的CO分電流密度。因此,通過改進型炭載體錨定金屬絡合物法,將小分子過渡金屬絡合物負載于疏水性炭載體中,通過炭化、酸洗等處理,能夠獲得分散性好、催化活性高的鎳-氮共摻雜催化劑,這為高活性電催化劑的制備提供了方法。
猜你喜歡
課堂內外·初中版(科學少年)(2025年1期)2025-02-28 00:00:00
課堂內外·初中版(科學少年)(2025年2期)2025-02-28 00:00:00
英語世界(2023年10期)2023-11-17 09:18:18
科學大眾(中學)(2019年3期)2019-05-17 10:04:30
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
汽車觀察(2018年10期)2018-11-06 07:05:26
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
