甲苯與甲醇側鏈烷基化制苯乙烯的研究進展
2021-09-15 00:46:46李修儀周金波黃劍鋒卜婷婷馬艷捷
石油煉制與化工 2021年9期
關鍵詞:催化劑
李修儀,周金波,黃劍鋒,卜婷婷,馬艷捷
(中國石油石油化工研究院蘭州化工研究中心,蘭州 730060)
苯乙烯作為重要的化工中間體,是橡膠和塑料行業最重要的原材料之一,主要用于生產聚苯乙烯(GPPS/HIPS、EPS等)、樹脂(SAN,ABS,UPR等)、丁苯橡膠(SBR)、涂料及絕緣材料等,用途十分廣泛。近年來,國內苯乙烯需求量不斷增長,進口量常年居高不下[1]。工業上生產苯乙烯主要采用乙苯脫氫法和環氧丙烷-苯乙烯聯產法。其中,采用乙苯脫氫法制得苯乙烯的產量占其總產量的90%以上[2]。
乙苯脫氫法制苯乙烯的反應過程為:在催化劑的作用下,乙苯脫除乙基上的氫生成苯乙烯。工業上典型的乙苯脫氫制苯乙烯技術主要有Lummus/UOP工藝和Fina/Badger工藝。乙苯脫氫反應過程為強吸熱且分子數增加的過程,需要較高的反應溫度和較低的反應壓力,因而對催化劑和反應器的要求均較高。同時,由于反應溫度較高,乙苯也會發生脫乙基或脫甲基反應,生成苯、甲苯、甲烷、乙烯和氫氣等副產物,因而產物需要分離提純。此外,原料乙苯要先由苯與乙烯烷基化制得,因此以該方法生產苯乙烯不僅工藝復雜、能耗高,而且要消耗高附加值的乙烯原料。
采用環氧丙烷-苯乙烯聯產(PO/SM)法生產苯乙烯,會聯產環氧丙烷。相較乙苯脫氫法,該工藝更加簡單、經濟,但后續分離過程復雜,并受環氧丙烷市場需求及利潤的限制,因而更適合于具有相關產業鏈的大型煉化企業。該技術工業上的典型應用有Halcon工藝。我國萬華化學公司于2019年宣布自主開發出高效綠色生產環氧丙烷-苯乙烯的成套技術,目前正在建設一套650 kt/a苯乙烯聯產300 kt/a環氧丙烷的工業裝置[3]。
鑒于上述兩種生產苯乙烯的工藝都存在一定的缺陷,研究人員不斷探索開發工業生產苯乙烯的新技術。其中,甲苯與甲醇進行側鏈烷基化反應可以制得苯乙烯,其反應方程如式(1)所示。

(1)
與傳統生產苯乙烯工藝相比,該技術路線可用煉油化工副產的大量甲苯和廉價的甲醇為原料,一步反應得到苯乙烯,原料廉價、工藝簡單、能耗低,是一種極具競爭力的苯乙烯制備技術。2007年美國Exelus公司的甲苯甲醇制苯乙烯技術(ExSyM工藝)完成中試試驗,相較乙苯脫氫工藝,其能耗更低,成本可降低約35%[4]。但是,由于缺乏高效的催化劑,該技術路線制備苯乙烯過程中甲醇利用率和苯乙烯收率均較低,因而至今仍無工業應用的報道。因此,開發適用于該技術路線的高效、高選擇性催化劑成為該領域研究的焦點。
1 甲苯側鏈烷基化反應的機理和熱力學
1.1 反應熱力學
甲苯與甲醇進行側鏈烷基化反應可以分為兩個步驟:一是甲醇脫氫轉化為甲醛,二是甲苯與甲醛發生側鏈烷基化反應生成苯乙烯。其反應過程的熱力學方程見式(2)~式(4)。
(2)
(3)
(4)
由式(2)和式(4)可知,甲醇脫氫反應會伴隨甲醇分解副反應,且二者均為強吸熱反應[5],室溫無法自發進行,升高溫度對其反應過程有利。但是,甲苯的側鏈烷基化反應為放熱反應,降低溫度對其反應過程有利。因此,需要選擇適宜的反應溫度,平衡甲醇脫氫與分解的競爭,使甲醇能夠脫氫生成甲醛,且甲醛作為烷基化試劑可與甲苯發生側鏈烷基化反應。溫度過低時,會抑制甲醇脫氫反應;溫度較高時,甲醇易發生分解副反應,降低其有效利用率,同時高溫也不利于甲苯與甲醛進行烷基化反應。而且,與甲醇脫氫反應相比,甲醇分解副反應的焓變較小,在熱力學上比前者更易發生。這正是甲醇利用率和甲苯轉化率均較低的原因。因此,需要開發對甲醇脫氫反應有高選擇性的催化劑,大幅降低其反應活化能,從而抑制甲醇分解副反應,控制反應沿主反應途徑進行。
1.2 反應機理
為了解甲苯與甲醇發生側鏈烷基化反應的機理,研究人員原位監測甲苯、甲醇及中間產物甲醛在催化劑上的吸附狀態。反應過程中,研究人員檢測到了吸附的甲醛,并對生成甲醛的量與甲苯側鏈烷基化反應的選擇性進行關聯,發現二者具有相關性[6]。因此,推斷甲苯與甲醇發生側鏈烷基化反應的過程為:首先,甲醇在催化劑表面的堿性中心脫氫生成甲醛;然后,甲醛作為烷基化試劑與甲苯上的甲基發生烷基化反應,生成苯乙烯。
Yashima等[7]對比了甲醛和甲醇分別與甲苯發生側鏈烷基化的反應過程,發現兩反應的選擇性相近,且甲醛與甲苯反應生成苯乙烯的收率更高,進一步證實了甲醛是真正烷基化試劑的合理性。Palomares等[8]與Rep等[9]使用原位紅外光譜研究甲苯和甲醇在堿性分子篩上的吸附和反應過程,剖析了甲苯與甲醇在堿金屬交換的X分子篩(KX,RbX,CsX)上發生側鏈烷基化反應的離子反應機理,如圖1所示。由圖1可知,甲醇首先在催化劑表面堿性中心上反應生成甲醛;甲醛作為烷基化試劑被吸附在催化劑表面的酸性中心上,形成碳正離子;同時,甲苯側鏈上的甲基與分子篩表面的堿中心([AlO-])作用而活化了C—H,使甲基上的碳被極化為碳負離子,苯環上的大π鍵電子作為給電子體與電子受體相互作用,使甲苯的苯環被催化劑酸性中心吸附;最后,極化的甲基碳負離子與甲醛碳正離子反應,生成苯乙烯并脫附。
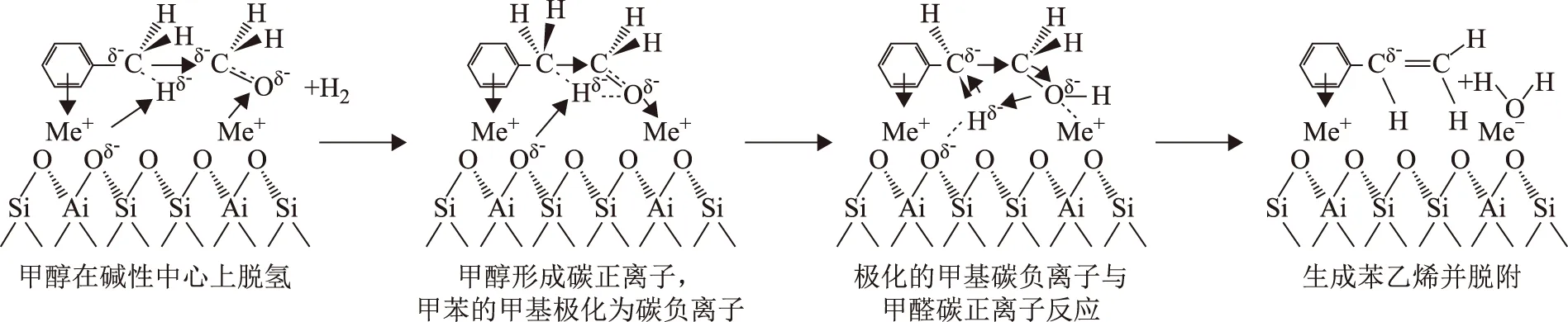
圖1 堿性分子篩上甲苯與甲醇進行側鏈烷基化反應的過程示意[9]Me為金屬K,Rb,Cs
但是,也有學者認為甲苯與甲醇進行的側鏈烷基化反應并不完全遵循上述離子反應機理。如Chen Huanhui等[10]通過對甲苯與甲醇側鏈烷基化反應進行同位素示蹤發現,甲苯與氘代甲醇(CD3OD)反應后,多數2D轉移到了甲苯的甲基上,只有很少量2D出現在苯乙烯和乙苯上,因此其提出了一種自由基反應機理:首先,甲苯裂解生成C6H5CH2·和H·;然后,C6H5CH2·與CH3OH反應生成2-苯基乙醇(C6H5CH2CH2OH);最后,2-苯基乙醇脫水生成苯乙烯。此外,通過量子化學計算可知,甲苯與甲醇發生氫氘交換的過程,發生自由基反應的活化能比發生離子反應的活化能更低。這也說明甲苯與甲醇發生側鏈烷基化反應時有部分自由基反應存在。
另外,對于該側鏈烷基化反應過程中乙苯的生成路徑也有爭議,傳統觀點認為產物中的乙苯是由苯乙烯發生連串反應加氫生成的[11],但也有學者認為乙苯是由苯乙烯和甲醇發生氫轉移反應生成的。Hattori等[12]和Alabi等[13]比較了苯乙烯分別與甲醇、氫氣的反應過程,以探究乙苯的生成路徑,發現苯乙烯與甲醇的氫轉移速率高于苯乙烯加氫,因而認為乙苯是由苯乙烯和甲醇在堿性催化條件下發生氫轉移反應生成的,而在該反應條件下苯乙烯加氫生成的乙苯量非常少。
2 催化劑對甲苯側鏈烷基化反應的影響
2.1 催化劑匹配酸/堿中心對甲苯與甲醇反應過程的影響
自Sidorenko等[14]發現甲苯與甲醇在堿金屬交換的X和Y分子篩上反應生成苯乙烯和乙苯后,研究人員進行了深入的研究[15-16],發現甲苯與甲醇在催化劑堿性中心上進行側鏈烷基化反應,但反應過程中需要催化劑的酸性中心吸附穩定苯環,以避免副反應發生。王文月等[17-18]通過研究X分子篩上甲苯與甲醇的吸附熱,發現催化劑的活性與其表面堿性中心數量、強度、分布并不成正比,而與強度、數量相匹配的酸/堿中心有相關性,因此推斷催化劑上相匹配的中強堿/弱酸中心是其具有高活性的關鍵,而強堿中心會催化苯乙烯生成乙苯,降低苯乙烯的選擇性。
Vasanthy等[19]用苯胺或苯酚分別毒化催化劑的堿/酸中心,發現當催化劑表面的酸中心或堿中心被完全毒化后,其催化甲苯側鏈烷基化反應的活性很低。當催化劑上的堿中心被完全毒化后,催化劑催化甲苯發生苯環烷基化反應,生成對二甲苯;當催化劑上的酸中心被完全毒化后,甲苯側鏈烷基化選擇性為零,而甲醇幾乎全部分解成CO和H2。Borgna等[20]引入酸/堿試劑毒化催化劑的堿/酸中心也得到相同的結論,證明甲苯與甲醇發生側鏈烷基化反應需要催化劑酸/堿中心協同作用。
因此,催化劑的酸/堿性質對甲苯側鏈烷基化反應的選擇性和甲醇反應路徑均有重要影響。催化劑酸性過強,甲苯會發生苯環烷基化或歧化反應,生成苯、二甲苯、三甲苯,甲醇則易脫水生成二甲醚;催化劑堿性過強,甲醇會分解成CO和H2,并促進甲苯發生連串反應生成乙苯,降低甲醇利用率和苯乙烯選擇性。甲苯和甲醇在酸/堿催化劑上可能發生的反應路徑如圖2所示。

圖2 甲苯和甲醇在催化劑表面酸/堿中心發生反應的主要路徑
2.2 催化劑電負性對甲苯側鏈烷基化反應的影響
由于分子篩堿性強弱與其骨架氧原子的局部電荷相關,而骨架氧原子的局部電荷又與其電負性(Sint)有關,因而學者們關聯催化劑的電負性與甲苯側鏈烷基化反應性能間的關系,以表征催化劑的性能。根據Sanderson電負性理論,多組分材料的電負性與其組成各元素的電負性相關,因此可以將電負性作為表征催化劑活性的特征之一。Giordano等[21]研究發現:當催化劑的電負性低于3.6時,催化劑對甲苯側鏈烷基化反應具有催化活性;而催化劑電負性大于3.6時,催化劑顯示對苯環烷基化有催化活性。然而,Kosova等[22]研究發現,分子篩不但要有合適的電負性,而且其骨架上相鄰Al原子要有合適的間距才能夠活化甲苯上的甲基。這說明,甲苯側鏈烷基化反應性能不僅與催化劑的電負性相關,也受分子篩骨架成鍵結構的影響。
2.3 催化劑空間結構對甲苯側鏈烷基化反應的影響
甲苯側鏈烷基化反應中,甲苯、苯乙烯和乙苯等反應物或產物的分子尺寸均較大,其中甲苯分子直徑約0.86 nm、動力學直徑約0.61 nm,因此要求催化劑的孔道必須足夠“寬敞”,以滿足反應物和產物擴散的要求。Itoh等[23]采用量子化學計算方法研究了甲苯的側鏈烷基化反應,發現除了酸/堿性質,催化劑的空間結構對該反應也有重要影響。不同分子篩的孔道結構如表1所示。由表1可知:ZSM-5,MOR,A,β分子篩的孔徑小于甲苯分子尺寸,限制了甲苯側鏈烷基化反應過程中過渡態的形成及反應產物的擴散,因而反應只能在其表面發生,催化劑活性低[13];X和Y分子篩具有大超籠結構(1.18 nm),孔口直徑也較大(0.74 nm),能夠提供足夠“寬敞”的反應場所,同時其酸/堿中心性質匹配、能夠協同,是較為理想的甲苯側鏈烷基化反應催化劑。

表1 不同類型分子篩的孔結構信息
3 甲苯側鏈烷基化反應的催化劑
用于甲苯側鏈烷基化反應的催化劑有分子篩類催化劑和非分子篩類催化劑。近年來,對于甲苯側鏈烷基化催化劑的研究,多圍繞采用不同方法對分子篩類催化劑改性,以調節其酸/堿匹配性,提高其催化活性和穩定性;同時,探索以多孔金屬氧化物、水滑石、炭材料等非分子篩類材料作為該反應催化劑的研究在不斷增多。
3.1 分子篩類催化劑的改性
3.1.1 金屬交換改性最初,堿金屬交換的X和Y分子篩被發現具有甲苯側鏈烷基化催化活性,但NaX和NaY分子篩對該反應的催化活性很低[24],必須進行改性才能提高其催化活性。用ⅠA族堿金屬(Li,K,Rb,Cs)對X和Y分子篩進行改性,發現分子篩催化活性與交換堿金屬的離子半徑相關,金屬離子半徑越大,催化劑活性越高。Cs改性X分子篩活性最高,Rb、K次之,而Li改性的X分子篩沒有甲苯側鏈烷基化催化活性,具有苯環烷基化活性。這是因為Li交換X分子篩表面以酸性中心為主,而其他堿金屬改性X分子篩表面具有不同強度的堿性中心。
同時,改性分子篩的匹配酸/堿性中心數量隨著其堿金屬含量(交換度)的增加而增多,其側鏈烷基化反應活性相應升高,但其側鏈反應選擇性(S苯乙烯+S乙苯)并不會直線提升。Li等[25]發現當Cs交換度大于32.3%時,甲醇脫氫生成甲醛反應為整個甲苯側鏈烷基化過程的關鍵步驟;而當Cs交換度小于32.3%時,甲苯的活化是其側鏈烷基化的關鍵步驟。Borgna等[26]發現,只有Cs交換度不低于40%時,NaCsX分子篩催化甲苯側鏈烷基化的選擇性才顯著提高;但另一方面,Cs交換度過高則會導致甲苯側鏈烷基化反應的選擇性降低。這是因為在Cs改性X分子篩上,催化甲苯側鏈烷基化反應的活性中心為O-Cs電子對(Oδ--Csδ+)幾何表面構型,而這種幾何構型只有在富Cs分子篩上才會形成,因而隨著Cs交換度的增大,CsX分子篩的催化活性提高;同時,甲醇的分解反應受晶格氧的堿度控制,增大Cs交換度有利于甲醇分解轉化為CO,而不是生成甲醛。因此,采用離子交換方法調節催化劑堿性需要控制其交換度,交換度過高或過低均不利于甲苯的側鏈烷基化反應。
王宇紅等[27]發現在X分子篩添加稀土元素La或Ce均能提高其活性,其中La的促進作用更明顯,同時La或Ce的添加都能提高催化劑的水熱穩定性。原因在于添加了La或Ce后催化劑的電負性增強,[AlO4]所帶電荷增加,給電子能力減弱,分子篩整體的堿性減弱,抑制了高溫反應過程中的水熱骨架脫鋁,提高了催化劑穩定性。
3.1.2 金屬氧化物改性Alabi等[13]在改性CsX的基礎上引入堿金屬氧化物Cs2O,發現Cs2O的引入能夠提高甲苯的轉化率,但會降低苯乙烯的選擇性。這是因為引入Cs2O后,CsX催化劑表面產生了明顯的強堿中心,促進甲苯甲基上的質子發生極化,有利于甲苯的轉化;但催化劑堿性增強也促進了甲醇分解,同時催化劑的酸性沒有變化,因而不利于苯乙烯的生成。這進一步說明了甲苯側鏈烷基化催化劑的堿性并不是越強越好。Han He等[28]研究發現,在催化甲苯側鏈烷基化反應中與骨架結合的Cs+和孔道中的Cs2O發揮的作用不同。其中,Cs+改變骨架氧的堿強度,起吸附和活化甲苯的作用;而Cs2O則促進甲醇有效轉化為甲醛。但是,若Cs2O過多則會加速甲醇的分解,反而降低側鏈烷基化選擇性。因而,骨架Cs+和Cs2O的協同作用對苯乙烯和乙苯的生成具有重要意義。
Hattori等[29]考察了Zn,Cu,Fe,Al等13種金屬的氧化物對CsX分子篩催化甲苯側鏈烷基化反應性能的影響,發現只有用ZnO改性CsX能夠促進甲醇轉化為甲醛并抑制苯乙烯氫化,對甲苯轉化率和苯乙烯選擇性的提高均有促進作用。
3.1.3 堿金屬鹽改性Han He等[30]在Cs2O/CsX上引入硼酸鹽,發現硼酸鹽的引入能夠明顯提高苯乙烯的選擇性,反應中苯乙烯選擇性可達17%(側鏈產物收率可達35.0%)。Wieland等[31]發現B的引入能夠中和催化劑表面較強堿性中心,抑制甲醇分解,進而提高側鏈烷基化選擇性。眾所周知,B原子不是富電子原子,其酸性來源不是B原子給出質子,而是B原子作為缺電子原子,能夠加合水分子中的氫氧根離子,進而釋放質子。此外,Li Peidong等[32]研究發現,在CsX催化劑中引入B元素能夠有效吸附和穩定甲醇脫氫生成的甲醛,并防止產物苯乙烯加氫轉化為乙苯。進一步研究發現,催化劑中B元素與堿性中心間的距離對甲苯側鏈烷基化反應具有重要影響,在CsX中摻混硼酸浸漬的氧化硅(B/SiO2)能夠顯著提高產物苯乙烯的選擇性。
利用非金屬原子的缺電子性質,在分子篩中引入其堿金屬鹽可生成穩定的配合物,以修飾催化劑的酸/堿性質。Wang Bin等[33]發現在負載K3PO4的CsX上,甲醇轉化率和甲苯側鏈烷基化的選擇性分別可達84.1%和92.8%,原因在于引入適量的K3PO4可以減少弱酸數量,增加中強堿數量。
類似于堿金屬的硼酸鹽、磷酸鹽,堿金屬碳酸鹽也能夠調變分子篩的酸/堿性質。林丹等[34]通過考察堿金屬碳酸鹽對KX和NaY催化劑活性的影響,發現堿金屬碳酸鹽與反應物作用可形成不同種類、數量的甲酸鹽,而甲酸鹽可使甲醇在較低溫度下脫氫轉為甲醛,從而提高甲苯的轉化率和側鏈烷基化產物的收率。但是,中間物種甲酸鹽對甲苯側鏈烷基化反應的作用還有爭議:King等[35]認為反應過程中形成的單齒甲酸鹽作為反應的一個過渡態參與了側鏈烷基化反應;Philippou等[36]認為反應過程中形成的中間體甲酸鹽不參與反應;而Hunger等[37]則認為表面形成的甲酸鹽物種是導致催化劑失活的原因。
3.1.4 分子篩改性的方法對X分子篩的改性除金屬交換和浸漬負載外,還有機械混合和固相反應法。Tope等[38]比較了不同改性方法對金屬硼酸鹽[ZrB2O5,Cu(BO2)2,LaB3O6等]修飾CsX催化劑對側鏈烷基化反應催化性能的影響,發現機械混合(CsX和金屬硼酸鹽混合均勻后焙燒)是金屬硼酸鹽改性CsX的較好方法。這是因為使用機械混合法對分子篩改性,改性物種只是覆蓋在分子篩的外表面,很難進入分子篩孔道內部;而催化劑外表面的部分酸中心被堿性物種中和,既可以促進甲醇脫氫生成甲醛,也可以減少苯環烷基化反應的發生[39]。
Wang Xiangsheng等[40]分別考察了KX與Al2O3,KY,KM,KZSM-5分子篩的二元混合物催化甲苯側鏈烷基化催化活性,發現苯乙烯的選擇性隨著第二分子篩組分中[AlO4]四面體局部電荷的增加而提高,其中,KX/KZSM-5二元分子篩的催化活性要遠優于單一分子篩的,側鏈烷基化產物的收率可達22%。堿性較弱而酸性較強的KZSM-5利于甲醇的活化,甲醇活化的中間體從KZSM-5擴散至KX,再與甲苯反應,進而提高了甲苯側鏈烷基化的反應活性[41]。
由于甲醇脫氫轉化為甲醛是甲苯側鏈烷基化反應的關鍵,Han Qiao等[42]將甲醇脫氫催化劑與CsX分子篩摻混得到復合催化劑,并將其用于甲苯甲醇側鏈烷基化反應。結果發現,甲醇脫氫催化劑的引入可以提高該反應產物苯乙烯和乙苯的收率。其中,引入Na2B4O7和CuO/SiO2甲醇脫氫催化劑時,復合催化劑催化甲苯側鏈烷基化反應產物的收率最高。此外,該研究還發現,復合催化劑中兩種組分的空間排布效應對其催化甲苯側鏈烷基化反應的活性和產物分布有重要影響。
此外,針對分子篩原子結構的特殊性,可以采用高溫氮化方法調節其酸/堿性質。氮化法使N原子取代分子篩骨架上的部分O原子進入骨架,由于N原子的電負性較O原子低,因而氮化處理能夠增強分子篩骨架的Lewis堿性。Tanabe等[43]對X分子篩催化劑進行氮化處理后發現,催化劑表面的堿性中心增加,甲苯側鏈烷基化反應的選擇性也隨著催化劑氮含量的增加而提高。于愛敏等[44]對用于甲苯側鏈烷基化反應的磷鋁分子篩進行氮化處理后,也得出相同的結論。需要指出的是,雖然氮化處理能夠溫和地調變催化劑酸/堿性質,但其調變幅度十分有限,原因在于微孔分子篩的結構穩定、有序,結晶度高,N原子難以大量替代O原子,因此氮化處理并非調變催化劑酸/堿性質的常用方法。
綜上所述,對X分子篩改性的目的均為改善催化劑的酸/堿性質,以形成更為匹配的酸/堿活性中心,促進甲苯與甲醇發生側鏈烷基化反應,抑制副反應。堿金屬、堿土金屬、稀土金屬以及非金屬硼酸鹽、磷酸鹽等的引入均對提高X分子篩的催化性能具有促進作用。除離子交換和浸漬法外,機械混合、高溫氮化等方法也可以改善X分子篩催化甲苯側鏈烷基化反應的性能,但改善效果有限。
3.2 非分子篩類催化劑
Wieland等[24,31]考察了不同材料作為甲苯側鏈烷基化反應催化劑的活性,發現具有一維、二維、三維孔結構的分子篩和炭材料均對該反應有催化活性,而堿金屬浸漬的Al2O3和MgO對該反應沒有催化活性。雖然MgO對甲苯側鏈烷基化反應沒有催化活性,但Jiang Nanzhe等[45]發現,若將MgO負載在以活性炭為模板劑的介孔Silicate-1分子篩上,則對該反應有催化活性。另外,Aziz等[46]發現介孔氧化硅納米粒子(MSN)對甲苯側鏈烷基化反應的催化活性比SiO2更高。這些研究從側面說明,催化材料的孔結構對其催化活性和催化反應過程均有重要影響;同時,材料酸/堿中心的匹配度也有較大影響,單純的介孔堿性金屬氧化物不具有催化活性。
Wang Bin等[47]以三嵌段共聚物普朗尼克F127為模板劑,以酚類化合物和甲醛為原料,采用蒸發誘導有機組裝法制備了新型Al-N固體酸/堿催化劑,并將其用于甲苯側鏈烷基化反應,發現側鏈反應產物(苯乙烯+乙苯)的總收率可達41.0%,遠高于用堿金屬交換分子篩催化的反應結果。分析其中原因,判斷合成催化劑表面酸/堿對的強度和間距均對甲苯與甲醇發生側鏈烷基化反應過程有利。
層狀復合氧化物鎂鋁水滑石由鎂八面體和鋁氧八面體構成,具有獨特的堿催化活性。Hao Chungao等[48]研究發現:類水滑石材料對甲苯側鏈烷基化反應中側鏈烷基化產物的選擇性達50.95%,其中苯乙烯選擇性為13.06%;而用磷酸鉀修飾該類水滑石材料,可將側鏈烷基化產物的選擇性提高至79.93%,其中苯乙烯選擇性為39.25%。這是因為磷酸鉀的加入調節了催化劑的酸/堿性質,使堿性中心的強度和數量更適宜,促進了苯乙烯的生成。Manivannan等[49]也考察了Mg/Al水滑石催化甲苯側鏈烷基化反應活性,甲苯的轉化率和側鏈烷基化產物的收率分別可達63.4%和31.9%。進一步研究發現,若用金屬離子Zn2+,Co2+,Ni2+,Cu2+等取代Mg2+后,其催化活性均不如Mg/Al水滑石[50];而用Ca2+取代Mg2+的Ca/Al水滑石與Mg/Al水滑石有相似的催化性能,因為Mg/Al水滑石和Ca/Al水滑石表面均有不同強度的堿性中心和弱酸中心。這也證明匹配的酸/堿性中心是甲苯與甲醇發生側鏈烷基化反應的必要條件。
3.3 催化劑的失活
對于甲苯側鏈烷基化反應,甲苯與甲醇發生側鏈烷基化生成苯乙烯和乙苯的同時,會有等物質的量的水生成。高溫下,水蒸氣分壓局部增大,會破壞分子篩結構,使分子篩骨架脫鋁,造成催化活性下降。研究表明,水熱脫鋁是導致分子篩結構變化、催化劑失活的主要原因[51]。另外,若脫附不及時,產物苯乙烯會聚合形成積炭,也會造成催化劑失活。對于積炭造成的催化劑失活,可以通過燒焦再生;而水蒸氣對分子篩造成的結構破壞是不可逆的,催化劑活性不能通過再生恢復。因此,提高催化劑穩定性的關鍵是改善分子篩骨架的水熱穩定性并抑制積炭的生成。對于水熱脫鋁造成的分子篩結構改變、活性下降,可通過高溫水熱處理、引入其他元素等措施,提高分子篩骨架在高溫水蒸氣下的穩定性。而對于積炭引起的催化劑失活,可通過調變催化劑表面的酸/堿性質,以減少積炭生成;通過引入介孔,來提高催化劑的容炭能力[52],從而減緩積炭對反應的影響。
4 結束語
與傳統苯乙烯生產工藝相比,甲苯與甲醇進行側鏈烷基化反應生產苯乙烯,工藝簡單、原料來源廣、成本低,是有競爭力的苯乙烯生產技術路線,其中催化劑是關鍵;但由于高效催化劑尚未開發成功,側鏈烷基化產物的收率較低,限制了其工業應用。
甲苯側鏈烷基化反應的催化劑主要以改性X分子篩為主,部分炭材料和水滑石材料也具有一定的側鏈烷基化催化活性,但效果并不理想。對X分子篩的改性方法主要有離子交換法、浸漬法、機械混合法、高溫氮化法等。堿金屬、堿土金屬、稀土金屬以及非金屬硼酸鹽、磷酸鹽等的引入對提高X分子篩的催化性能均有促進作用;而機械混合法、高溫氮化法對改善X分子篩的催化性能效果有限。
對甲苯與甲醇進行的側鏈烷基化反應,催化材料的孔結構對其催化活性和反應過程均有重要影響;催化劑具有匹配的酸/堿中心是甲苯側鏈烷基化發生的必要條件,單純的介孔堿性金屬氧化物不具有催化活性。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
