釩改性對鐵基脫硝催化劑活性及抗堿性能的影響
2021-09-22 07:32:20蔡思翔
人工晶體學報 2021年8期
關鍵詞:催化劑
李 悅,姜 宏,蔡思翔
(1.海南大學材料與科學工程學院,海口 570228; 2.海南大學,海南省特種玻璃實驗室,海口 570228; 3.海南大學,南海海洋資源利用國家重點實驗室,海口 570228)
0 引 言
氮氧化物(NOx)的排放對全球環境及人體健康造成了巨大危害,NH3-SCR技術是當前應用得最為廣泛的氮氧化物減排技術之一[1-3]。鐵基催化劑以其突出的抗硫性能、優異的熱穩定性、良好的中高溫催化活性以及價格低廉、綠色環保等優勢受到廣泛關注[4-5]。然而鐵基催化劑仍存在著低溫活性差以及溫度窗口窄的問題,因此許多研究者以晶相調控或表面結構調變的方法來改善催化劑的氧化還原循環和酸循環,以提升其低溫SCR活性并拓寬溫度窗口[6-8]。此外,摻雜其他金屬氧化物、采用多元復合催化體系改性等方式也能對催化劑進行有效改性。有研究發現向Fe-Ti尖晶石中摻入V/Mn后,NO轉化率明顯增加,特別是在200~300 ℃時,向Fe-Ti尖晶石加入V對SCR反應的影響比加入Mn明顯得多[9]。Zhang等[10]的研究也發現加入少量V形成的鐵釩混合氧化物Fe-V-Ox可以提高催化劑的氧化還原性能和催化活性。Mu等[11]制備的釩摻雜Fe2O3催化劑由于氧化還原能力和表面酸性的提高,在175~400 ℃的寬溫度范圍內可達90%以上的NOx去除率,且同時展現出良好的N2選擇性以及抗H2O/SO2性能。近年來,釩酸鐵(FeVO4)作為一種具有優異的低溫催化活性、寬工作溫度窗口以及突出的抗硫性能的金屬釩酸鹽,受到越來越多研究者的關注。釩酸鐵與傳統的釩氧物種相比,具有較高的熱穩定性[12],因此相較無法規避釩生物毒性的傳統VWT催化劑而言,更具綠色環保性。Liu等[13]將釩酸鐵負載到不同載體上,發現FeVO4/TiO2催化劑在所有催化劑中具有最佳活性。高分散的FeVO4的活性相只存在于催化劑表面,而并未進入載體TiO2的晶格結構中。且在FeVO4相表面富集的VOx物種是真正的催化活性相,同時催化劑中存在大量與反應物在催化劑表面的吸附和活化有關的表面缺陷。
堿金屬及堿土金屬化合物(主要為Na2O、K2O、MgO、CaO等)大量存在于以化石、生物質等為燃料的工廠或生活垃圾焚燒過程等排放的煙氣中,主要通過對催化劑表面的酸性位點和活性中心的影響來破壞催化反應中的酸循環和氧化還原循環[14-15]。另外,對于金屬氧化物基催化劑,堿金屬對催化劑的毒害影響一般大于堿土金屬,而堿金屬中鉀的毒害作用又最強[16-17]。然而當前針對鐵基催化劑抗堿性能的研究非常少,此外其低溫性能也亟待改善。因此設計開發具有低溫活性好、抗堿中毒性能佳的新型鐵基脫硝催化劑并對其進行相關研究具有重大意義。
本文通過引入V物種制備低溫活性優異且溫度窗口較寬的氧化鈦負載的鐵釩催化劑(FeV/TiO2),并通過一系列性能及表征測試研究了V改性對催化劑活性及抗堿性能的影響。
1 實 驗
1.1 實驗材料
銳鈦礦型TiO2、Fe(NO3)3·9H2O、NH4VO3、草酸、氨水,均購于國藥集團化學試劑有限公司,去離子水實驗室自制。
1.2 催化材料制備
對于負載型鐵釩催化劑的制備[13],首先將一定量的草酸超聲溶解于50 mL去離子水中形成草酸溶液,向上述草酸溶液中加入一定量的偏釩酸銨并攪拌使之完全溶解,隨后再加入一定量的Fe(NO3)3·9H2O(nFe∶nV=1∶1),然后加入銳鈦礦型二氧化鈦載體。攪拌一段時間后在50 ℃下旋蒸至蒸干。置于100 ℃的烘箱中過夜干燥,將干燥后的產物置于馬弗爐中500 ℃下煅燒5 h,將制得的催化劑標記為FeV/TiO2。此外,非負載的鐵釩復合氧化物也被用相同的方法制備(除加入載體外),并標記為Fe0.5V0.5Oδ(nFe∶nV=1∶1);同時制備負載型氧化鐵催化劑作對比。將一定量的Fe(NO3)3·9H2O溶解于100 mL去離子水中,加入一定量的銳鈦礦型二氧化鈦載體并在室溫下攪拌使之混合均勻,向上述混合溶液中加入適量的氨水,調節pH值至10,室溫下連續攪拌3 h,用去離子水過濾洗滌,置于100 ℃的烘箱中過夜干燥后在馬弗爐中500 ℃下煅燒5 h,將制得的催化劑記為Fe2O3/TiO2。上述催化劑的負載量均為10%(質量分數)。
堿金屬中毒催化劑的制備過程如下:將一定量的硝酸鉀溶解于25 mL去離子水中,攪拌使之溶解,將制備的新鮮催化劑加入上述硝酸鉀溶液,在50 ℃下旋蒸至蒸干,干燥并煅燒(500 ℃,5 h)后制得鉀中毒的催化劑,并將上述催化劑標記為K-Fe2O3/TiO2、K-FeV/TiO2、K-Fe0.5V0.5Oδ,其中浸漬負載上的K2O的質量分數為1%。
1.3 催化材料表征
采用日本Rigaku公司D/Max-RB型X射線衍射(XRD)光譜儀來表征催化劑的晶體結構,掃描范圍10°~80°,掃速8(°)·min-1,測試電壓40 kV,電流40 mA,λ=0.154 18 nm;采用英國Jobin Yvon公司HR-800型拉曼(Raman)光譜儀測試催化劑表面分子鍵的振動情況,以532 nm的激光光源作為激發光源;利用美國Micromeritics公司ASAP 2460型氮氣物理吸附(BET)儀測試催化劑的比表面積信息,測試前在300 ℃下將樣品在脫氣系統(VacPrep 061)中預先脫氣12 h;使用美國Agilent公司Cary 5000型紫外-可見-近紅外(UV-Vis-NIR)分光光度計表征催化劑分子結構及電子轉移相關信息,使用BaSO4作為參考,通過漫反射光譜(DRS spectra)記錄樣品在200~800 nm范圍內的吸光度;采用美國Micromeritics公司Auto Chem HP 2950型全自動化學吸附儀進行氨氣程序升溫脫附(NH3-TPD)及氫氣程序升溫還原(H2-TPR)實驗,以測試催化劑表面酸性及氧化還原性能。具體操作為先稱取80 mg催化劑,以10 ℃/min的升溫速率在N2氣氛下升溫至300 ℃保持30 min進行預處理。結束預處理并降溫至100 ℃后,通入H2/NH3,進行程序加熱升溫至900 ℃。
1.4 催化性能測試
催化劑催化活性的測試在固定床反應器中進行(測試所用石英管內徑為7 mm)。具體測試條件為:NO、NH3氣體濃度為5.0×10-4,O2體積分數為5%,并以N2作為載氣,氣體總流量設置為260 mL·min-1左右,反應氣體體積空速為40 000 h-1。利用FT-IR光譜分析儀(Thermo Fisher Scientific)對進出口反應氣NOx、NH3、N2O的濃度進行檢測。催化劑的NO轉化率和N2選擇性通過以下方程計算:
(1)
(2)
2 結果與討論
2.1 催化劑性能分析
如圖1為催化劑的NOx轉化率隨測試溫度變化的曲線。引入V后形成的FeV/TiO2催化劑相較于Fe2O3/TiO2催化劑,其SCR活性顯著提升,溫度窗口大大拓寬,在210~420 ℃具有90%以上的NOx轉化率。對于堿中毒催化劑,K-FeV/TiO2在240~370 ℃溫度范圍內仍能保持80%以上的催化活性,而K-Fe2O3/TiO2則嚴重失活,甚至在高溫下完全失活。因此,引入V對催化劑的活性及抗堿金屬中毒性能具有明顯的提升效果。此外,還測試了催化劑的N2選擇性(見圖2),發現Fe2O3/TiO2催化劑在中毒前后的整個測試溫度范圍內均能保持95%以上的選擇性,FeV/TiO2與之幾乎相同但在高溫段選擇性略有下降,K-FeV /TiO2催化劑在360 ℃以上的高溫段下降較為明顯。
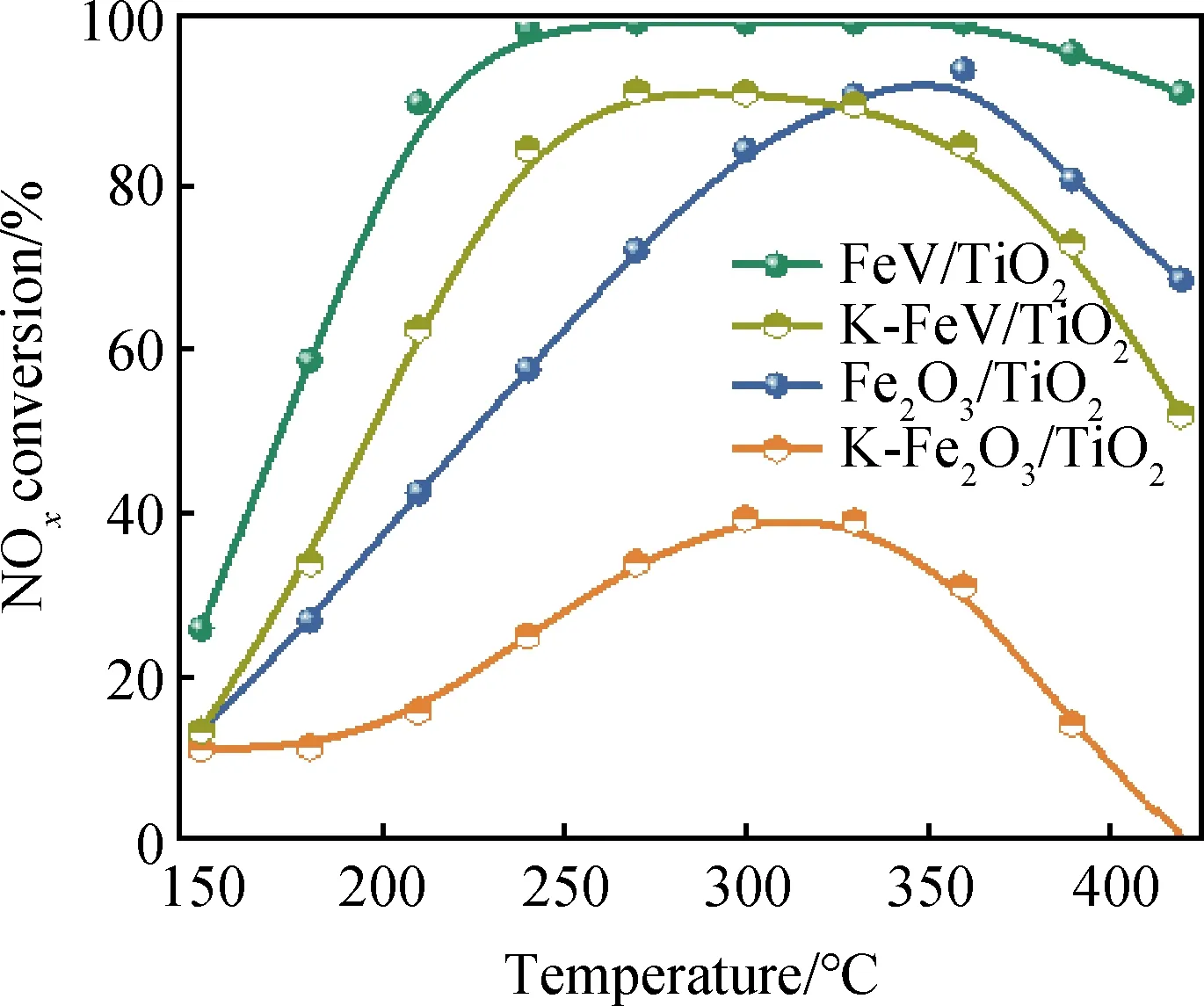
圖1 不同催化劑的NOx轉化率Fig.1 NOx conversion over different catalysts
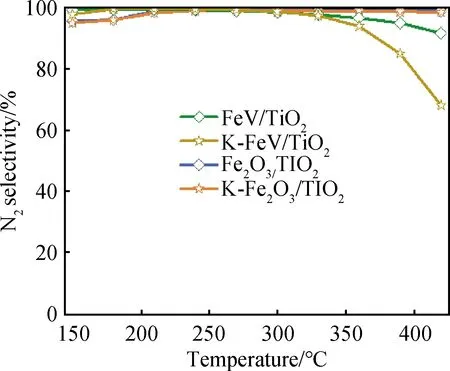
圖2 不同催化劑的N2選擇性Fig.2 N2 selectivity over different catalysts
2.2 催化劑結構分析


圖3 (a)TiO2負載型催化劑和(b)非負載型催化劑的XRD圖譜Fig.3 XRD patterns of (a) TiO2 supported catalysts and (b) unsupported catalysts
從Raman圖譜可以看到(見圖4(a)),出現在143 cm-1、394 cm-1、512 cm-1和635 cm-1處的峰主要是銳鈦礦型二氧化鈦的特征振動峰[5]。K-Fe2O3/TiO2催化劑中歸屬于TiO2的Eg峰(143 cm-1)的強度相較于Fe2O3/TiO2的變弱,且發生藍移(向高波數移動),這說明K可能與TiO2產生了鍵合作用,導致Ti—O鍵變得穩定而不易發生斷裂,從而不易被活化;而FeV/TiO2催化劑中相應的峰則只是稍許藍移,并未發生明顯變化,說明K中毒對其幾乎無影響[15]。同時觀察非負載鐵釩復合氧化物的Raman圖譜(見圖4(b))可以發現,非負載Fe0.5V0.5Oδ催化劑的拉曼特征峰主要顯示為FeVO4相(320 cm-1、382 cm-1、750 cm-1、822 cm-1以及911 cm-1處)[11,20]。

圖4 (a)TiO2負載型催化劑和(b)非負載型催化劑的Raman圖譜Fig.4 Raman spectra of (a) TiO2 supported catalysts and (b) unsupported catalysts
此外,由表1中BET測試的結果可以發現,引入V后的FeV/TiO2催化劑相較于Fe2O3/TiO2催化劑,其比表面積顯著增大,且在K中毒后仍具有較大的比表面積,從而可為NH3-SCR反應提供更多的活性位點[15]。

表1 不同催化劑的比表面積Table 1 Specific surface area of different catalysts
UV-Vis-NIR的測試結果顯示(見圖5),Fe2O3/TiO2在547 nm處出現了對應于Fe3+的吸收帶[21],而對于FeV/TiO2催化劑,其歸屬于Fe3+的吸收帶出現在512 nm處,較Fe2O3/TiO2發生紅移,說明其Fe3+的化學環境由于Fe與V之間強電荷相互作用而發生了明顯變化[22]。同時可以觀察到,Fe2O3/TiO2在742 nm左右處出現了屬于α-Fe2O3的一個典型負帶,而在FeV/TiO2催化劑中并未出現,這也說明了后者的分散性更好。此外,這兩種催化劑在K中毒后均未發生明顯變化。
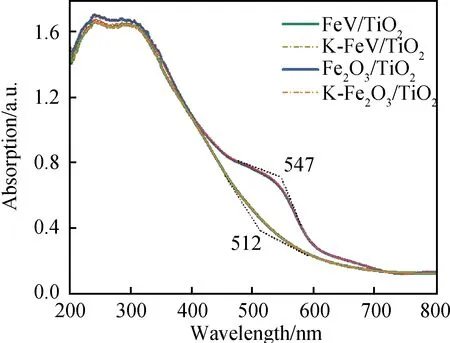
圖5 不同催化劑的UV-Vis-NIR圖譜Fig.5 UV-Vis-NIR DRS spectra of different catalysts
2.3 催化劑表面酸性及氧化還原性分析
如圖6中的NH3-TPD測試結果所示:與Fe2O3/TiO2相比,FeV/TiO2催化劑的NH3吸附容量大大增加(+55.61%),表明其酸性大大增強;中毒后,相較于K-Fe2O3/TiO2催化劑酸量的大大損失(NH3吸附容量下降了67.42%),FeV/TiO2催化劑在堿中毒后仍能保持相當的酸量,從而保證了NH3-SCR反應中反應氣體NH3在催化劑表面的吸附,這也是其在中毒前后均能表現出較高的反應活性的重要原因[23]。
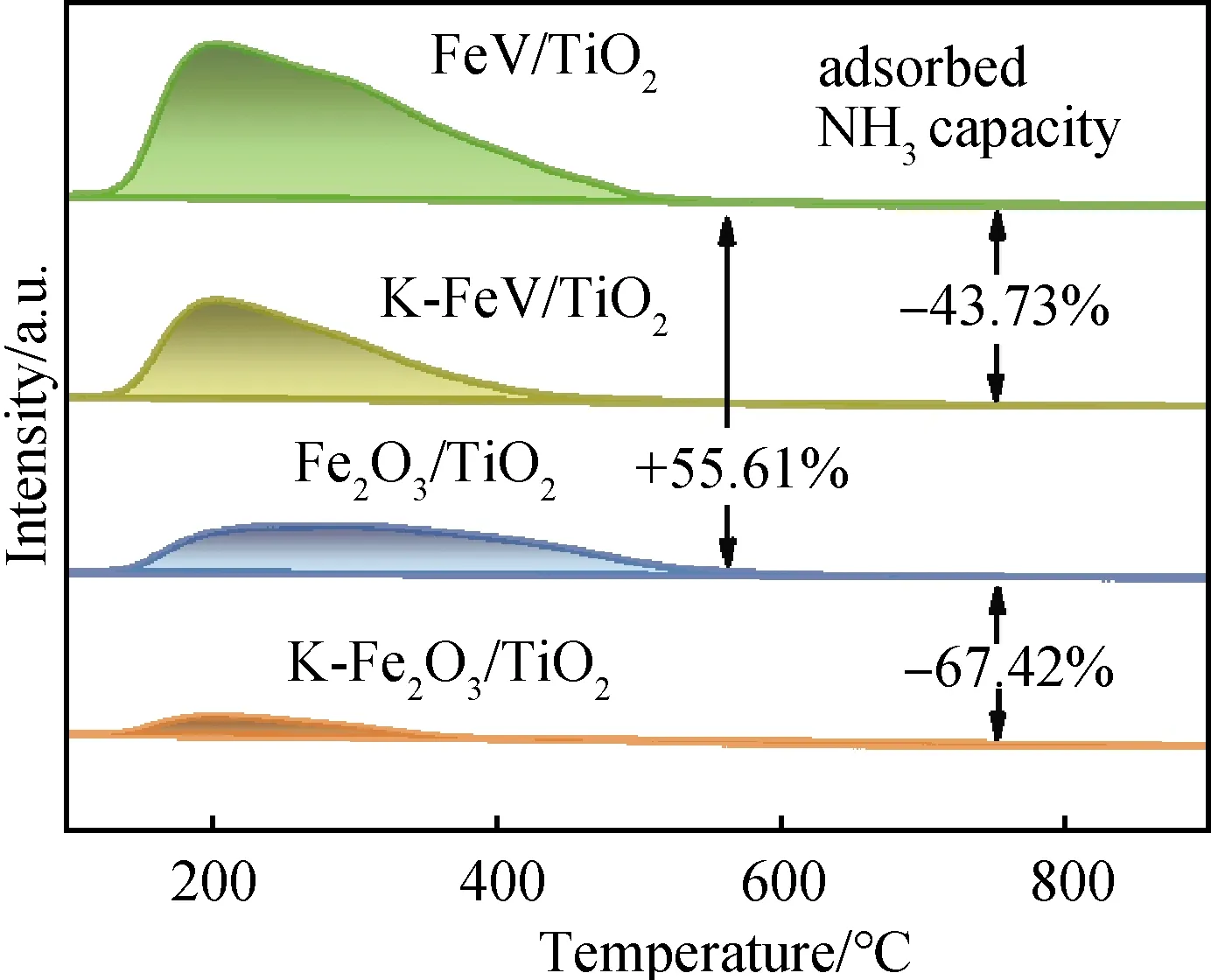
圖6 NH3-TPD-MS中不同催化劑的NH3的MS曲線Fig.6 MS signal of NH3 from NH3-TPD-MS profiles for different catalysts
催化劑的氧化還原能力也是影響其催化活性的重要因素。從圖7顯示的H2-TPR測試結果可知,Fe2O3→Fe3O4、Fe3O4→FeO、FeO→Fe的連續還原過程在Fe2O3/TiO2催化劑中所對應的還原溫度分別為324 ℃、363 ℃和426 ℃[5,24]。而在K中毒后分別向高溫區域移至381 ℃、418 ℃和514 ℃。在FeV/TiO2中,出現在398 ℃的還原峰為Fe3+→Fe2+與V5+→V4+的共還原峰[25],其后在更高溫度出現的還原峰則對應于Fe2+→Fe與V4+→V3+的共還原峰[26]。與Fe2O3/TiO2相比,其還原峰向高溫段有所轉移,這可能是由于引入V后形成的FeVO4物種抑制了Fe2O3的過度氧化;此外,其尖銳的峰型也說明了引入V后,活性組分在載體上的分散性大幅提高[23]。同時FeV/TiO2及其K中毒的K-FeV/TiO2的還原峰面積較Fe2O3/TiO2及K-Fe2O3/TiO2更大,這說明其H2消耗量較高,也即其氧化還原性顯著提升。結合紫外結果及上述對催化劑結構的分析可知,這可能是由于Fe、V之間發生了電荷轉移而產生的強電荷相互作用,以及FeVO4與Fe2O3間的電子相互作用[10-11]。
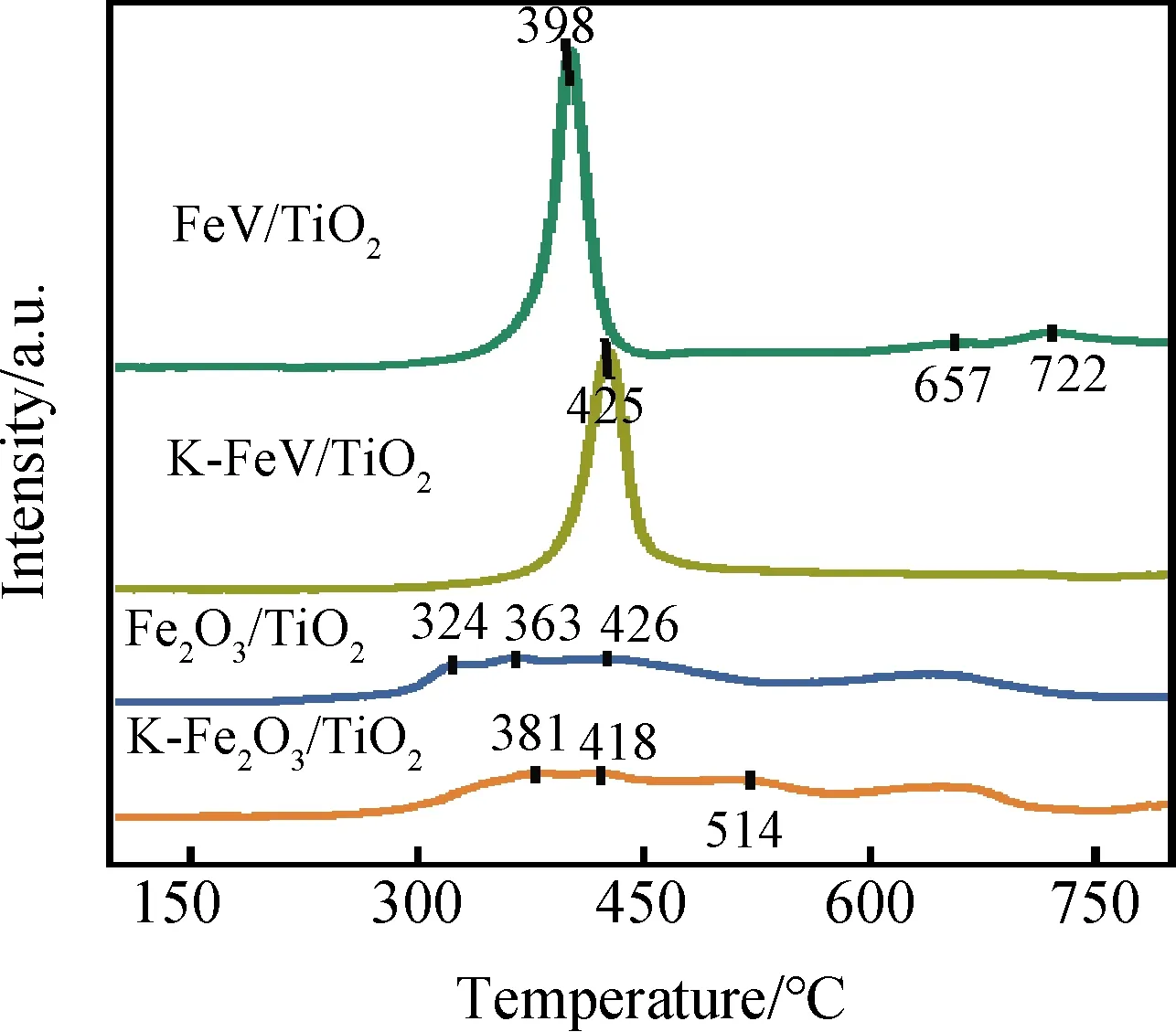
圖7 不同催化劑的H2-TPR曲線Fig.7 H2-TPR profiles of different catalysts
3 結 論
通過引入V制備了FeV/TiO2催化劑,與未改性的Fe2O3/TiO2催化劑相比,其低溫活性得到顯著提升且溫度窗口大幅拓寬,堿中毒后,FeV/TiO2催化劑在240~370 ℃內保持80%以上的催化活性。通過對催化劑結構性質和表面理化性質的表征,發現引入V后,催化劑呈現出以FeVO4為主相并伴有少量Fe2O3的狀態,且其結構幾乎不受K中毒的影響。催化劑中存在大量酸性位點而具有較強的表面酸性,Fe、V物種間的強相互作用也大大提升了其氧化還原性,因此催化劑具有優異的低溫活性和較寬的溫度窗口。此外,催化劑的結構幾乎不受堿中毒的影響,且在K中毒后仍能維持較強的表面酸性,從而使其具有良好的抗堿金屬中毒性能。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
