界面化學對鎳錳酸鋰正極材料電化學性能影響的研究現狀及分析
2021-11-14 08:59:56廖文俊
無機鹽工業 2021年11期
周 蘭,李 旺,廖文俊
(上海電氣集團股份有限公司中央研究院,上海200070)
近年來,對能源的巨大需求激發了高能量密度鋰離子電池材料的發展,尤其是電動汽車領域,目前商業化的鋰離子電池仍無法滿足電動汽車對電池低成本及高能量密度的要求。尖晶石型的LiNi0.5Mn1.5O4正極材料因其價格低廉、對環境無污染、安全性能好、高能量密度等優點而成為當今鋰離子電池正極材料研究的熱點之一。其理論放電比容量可達146.7 mA·h/g,電壓平臺高達4.8V,具有超高的能量密度(650 W·h/kg)及功率密度,目前已經引起廣泛關注,被認為是未來鋰離子電池發展中最有前景的鋰離子電池正極材料之一[1-3]。本文從LiNi0.5Mn1.5O4正極材料的結構及脫嵌機理入手,對近年來國內外在LiNi0.5Mn1.5O4正極材料和電解液方面的研究進展進行總結和分析,著重探討了界面化學對LiNi0.5Mn1.5O4正極材料電化學性能的影響情況,特別是近幾年圍繞LiNi0.5Mn1.5O4正極材料表面和電解液界面提出的新方法和新思路,并對今后的研究和發展趨勢進行了展望。
1 LiNi0.5Mn1.5O4正極材料的結構及脫嵌機制
LiNi0.5Mn1.5O4存在非化學計量比無序和化學計量比有序兩種尖晶石結構,分別對應Fd3m和P4332兩種空間群,如圖1所示[4],在Fd3m中,Ni和Mn原子在進行占位時,是隨機分布在16d位置,Li原子位于8a位置,O原子位于32e位置;而在P4332中,Ni原子和Mn原子有序地排列在鎳錳酸鋰晶格結構中,Ni和Mn原子分別占據4a和12d位置,Li原子占據8c位置,O原子分別占據24e和8c位置。
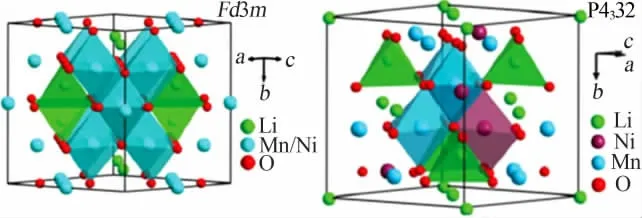
圖1 尖晶石型正極材料LiNi0.5Mn1.5O4結構示意圖[4]Fig.1 Structural diagram of spinel LiNi0.5Mn1.5O4 cathodes[4]
P4332有序型結構LiNi0.5Mn1.5O4中的Mn為+4價,在實際制備過程中,可以通過改變煅燒工藝或摻雜改性等方式向材料中引入微量Mn3+,為了保證材料的電中性,材料結構中會出現氧空缺,使得材料的空間群由P4332逐漸變化為Fd3m,這種結構的改變也被稱為LiNi0.5Mn1.5O4材料的無序化過程,無序程度不同Mn3+的含量也不同。通常認為,Fd3m型的LiNi0.5Mn1.5O4材料相比于P4332型具有更優異的倍率性能,這與晶胞參數大小及鋰離子的擴散系數有關,無序型Fd3m結構更有利于Li+和電子的傳輸,因而具有更好的導電性。有序型和無序型LiNi0.5Mn1.5O4可以通過XRD、紅外等多種方式判定,其中充放電曲線是最典型的判定方式之一,如圖2所示[5]。

圖2 有序型與無序型尖晶石LiNi0.5Mn1.5O4正極材料充放電曲線對比圖[5]Fig.2 Comparison diagram of charge and discharge curves of ordered and disordered LiNi0.5Mn1.5O4 spinel cathodes[5]
對于P4332型有序LiNi0.5Mn1.5O4材料,Mn完全為+4價,Ni為+2價,充放電過程中,只在4.7 V左右出現了單一電壓平臺,對應Ni2+/Ni4+的氧化還原反應過程;而對于Fd3m型無序LiNi0.5Mn1.5O4材料,在4.6 V左右和4.8 V左右出現了兩個平臺,分別對應Ni2+/Ni3+和Ni3+/Ni4+兩個過程,且由于微量Mn3+的存在,在4.0 V附近出現了Mn3+/Mn4+的小平臺[6]。
2 表面和界面化學
對于鎳錳酸鋰電池來說,各種副反應,包括高電位下電解液的分解、電極的腐蝕、金屬Mn、Ni的溶解及高活性Ni4+對電解液反應的催化等問題會導致活性材料的流失及電池阻抗的增大,最終使得電池庫倫效率降低、循環性能變差。而對于鋰離子電池來說,電子遷移和鋰離子擴散通常發生在活性材料表面以及與其他物質組成的各種界面之間,因此通過改善LiNi0.5Mn1.5O4材料的表面和界面化學對于提高鎳錳酸鋰電池的電化學性能有著至關重要的作用。
2.1 LiNi0.5Mn1.5O4表面化學
2.1.1 晶面取向
對于LiNi0.5Mn1.5O4材料來說,不同的晶面取向,不僅決定了其晶體形態的不同,更對其電化學性能有著重要的影響[7-13],這主要與Li+在LiNi0.5Mn1.5O4中擴散的各向異性動力學有關。例如HAI等[7]曾報道過板面形狀和八面體形狀的LiNi0.5Mn1.5O4,并對其不同的化學擴散系數進行了深入研究,結果表明八面體結構的LiNi0.5Mn1.5O4相比于板面結構的LiNi0.5Mn1.5O4則具有更高的容量和更好的倍率性能,這主要是八面體結構的LiNi0.5Mn1.5O4中對應的(111)晶面相比于板面結構的LiNi0.5Mn1.5O4中對應的(112)晶面具有更高的Li離子擴散系數。DENG等[8]利用固相法分別制備了P摻雜的八面體和截角八面體的LiNi0.5Mn1.5O4材料,如圖3所示。通過不同比例P摻雜向材料中引入不同量的Mn3+而使得LiNi0.5Mn1.5O4無序度增加的同時也對晶面形態產生了影響,如LiNi0.5-xP2xMn1.5-xO4中x分別為0和0.005時,晶體形態為正常的八面體形狀,僅具有(111)晶面,而當x逐漸增大為0.01和0.02時,晶體形態變為截角的八面體形狀,并伴隨著(111)晶面的減少及(100)晶面的增多。電化學結果表明,LiNi0.5Mn1.5O4材料摻雜P后材料的無序度增加,由于無序型Fd3m結構更有利于Li+和電子的傳輸,而使得材料的電化學性能大大提升;但隨著P摻雜量的進一步增加,晶體形態也發生了轉變,(100)晶面不斷增多而(111)晶面不斷減少,這時晶面取向開始占據主導地位,這時(111)晶面的大幅度減少會對Li+擴散產生不利影響,因此隨著P摻雜比例的進一步提高,材料的電化學性能下降。當然,CHEMELEWSKI等[9]也在文獻中進一步證實(111)晶面與電解液之間具有更優異的界面穩定性。但是,近年來也有一些其他觀點出現,CHEN等[13]通過液相可控法合成出了具有(111)晶面外還同時擁有(001)、(110)、(113)、(103)等晶面的類球形截角多面體結構,相較于僅有(111)面的八面體結構,這種截角多面體結構使得LiNi0.5Mn1.5O4材料具有更優異的容量保持率和倍率性能,這種性能的提升被歸功于(111)面的減少和(110)面的增加,CHEN等[13]認為(110)面更有利于鋰離子擴散。不僅如此,(111)面的減少還能有效抑制Mn的溶解,從而避免由于Mn3+的溶解而導致Jahn-Teller效應的發生,大大提高了材料的穩定性。LIU等[14]通過微波水熱法合成出具有(100)面的截角八面體結構的鎳錳酸鋰材料,并表現出相當不錯的倍率性能及長 循 環 性 能,因 此LIU等[14]認 為(100)面 相 較 于(111)面更有利于LiNi0.5Mn1.5O4材料電化學性能的提升。盡管如此,對于尖晶石體系來說,究竟是(111)面還是(100)面對電化學性能提升作用大仍存在爭議。筆者認為之所以會出現這兩種觀點,是因為對于LiNi0.5Mn1.5O4材料來說,電化學性能的好壞不僅與晶面取向有關,同時還與顆粒形貌、陽離子Ni2+和Mn4+在晶格中的排序、巖鹽相LixNi1-xO雜質的形成、Mn3+的含量、氧缺陷等眾多因素有關,在設計材料的多面體結構過程中,這些因素中的一項或多項也在跟著發生改變,從而無法準確判斷出電化學性能的好壞是否只是因為晶面結構的變化所導致。

圖3 高倍透射電鏡圖(a1~d1,a2~d2)[8]Fig.3 HRTEM images(a1~d1,a2~d2)[8]
2.1.2 顆粒形貌
LiNi0.5Mn1.5O4合成方法主要有固相法、共沉淀法、溶膠凝膠法、噴霧熱解法、水熱/溶劑熱法與熔鹽法等。不同的合成路徑、特定添加劑和表面活性劑將直接影響晶體生長,形成不同形貌。與微米級的LiNi0.5Mn1.5O4顆粒相比,納米尺寸的LiNi0.5Mn1.5O4具有更優異的倍率性能,但從循環穩定性的角度來說,納米顆粒雖然能夠有效縮短Li+的擴散路徑,提高材料的倍率性能,但其形成的大比表面會大大增加與電解液的接觸面積,加劇與電解液的副反應并增加過渡金屬離子的溶解,因此亞微米尺寸結合三維或多孔結構等特殊形貌的LiNi0.5Mn1.5O4材料目前研究的更多,效果也更好。LIU等[15]通過靜電紡絲結合退火處理的方法制得直徑為200~500 nm、長度為幾十微米的纖維結構,這種三維結構可以實現充放電過程中Li+和電子的快速傳輸,具有良好的電化學性能,在0.5C時,其初始放電容量為133 mA·h/g,30次循環后容量保持率超過94%。HAO等[16]采用低溫兩步水熱法成功得到亞微米尺寸的八面體形貌材料,表現出良好的倍率性能,1C時其放電比容量接近130 mA·h/g,5C時比容量仍接近100 mA·h/g。
2.1.3 表面元素分布
研究表明LiNi0.5Mn1.5O4表面的元素分布對其電化學性能也有很大影響,隨著尖晶石LiNi0.5Mn1.5O4中Mn3+含量的增多,通過歧化反應產生的Mn2+溶解到電解液中的量也會增多,因此降低LiNi0.5Mn1.5O4表面Mn3+的含量可以有效減少Mn2+向電解液中的溶解,然而Mn3+對LiNi0.5Mn1.5O4電化學性能的影響一直存在爭議。羅英等[17]研究發現通過優化Mn3+含量,控制材料形貌可以實現高性能LiNi0.5Mn1.5O4材料的制備。XIAO等[18]通過原子模擬指出,Mn3+的形成促進了Ni-Mn位點的無序排列,這種無序排列更有利于Li+的傳輸,尤其是大倍率充放電的情況下表現更為明顯。而且,由于Mn3+含量的增加所帶來的電子跳躍使得無序(Fd3m)尖晶石LiNi0.5Mn1.5O4的室溫電導率比有序(P4332)高出2.5個數量級,這對高溫下容量保持率是有益的。未來還需要進一步優化出Mn3+的最適比例,以此來平衡Mn3+對LiNi0.5Mn1.5O4電化學性能的雙重影響。
除了上述Mn元素影響外,Ni元素對LiNi0.5Mn1.5O4材料的電化學性能影響也很大。LIU等[19]研究發現,55℃下循環后的LiNi0.5Mn1.5O4電極出現了材料的粉化現象,歸咎于Ni促進了電解液的氧化分解,并在正極表面形成了鈍化層(CEI),從而阻礙了Li+的擴散,最終影響了循環性能。而Cr摻雜后的LiNi0.5-xCr2xMn1.5-xO4(0≤2x≤0.8),由于Cr3+部分取代了Ni2+和Mn4+,使得材料表面跟電解液接觸的Ni含量減少,這有利于電極/電解液的界面穩定性,降低了表面的阻抗,提高了鎳錳酸鋰材料的循環性能。LEE等[20]選用Cr-,Fe-和Ga-取代Ni-制備LiMn1.5Ni0.5-xMxO4(M=Cr,Fe,Ga)材料,也得出類似的結論。他們認為摻雜后的材料具有更穩定的電極/電解液界面,可以顯著提高材料的循環和倍率性能。原因是這幾種摻雜離子具有一定的自偏析效應,TOFSIMS深度測試結果如圖4所示。由圖4可以看出[20],與Ni相比,摻雜離子更容易富集在材料表面而造成表面貧Ni的狀態,進而有效抑制了電解液的氧化分解。

圖4 LiMn1.5Ni0.5-xMxO4(M=Cr,Fe,Ga;x=0、0.08)樣品的飛行時間二次離子質譜(TOF-SIMS)深度剖析分布圖[20]Fig.4 Time of flight secondary ion mass spectrometry(TOF-SIMS)depth profifiles of the LiMn1.5Ni0.5-xMxO4(M=Cr,Fe,Ga;x=0、0.08)samples[20]
2.1.4 表面結構的轉變
充放電過程中表面結構的變化很難直觀表征出來,因此關于表面結構的轉變對LiNi0.5Mn1.5O4電化學性能的影響鮮有報道。LIN等[21]通過STEM發現,與沒有充放電的原始LiNi0.5Mn1.5O4材料相比,充電到4.9V的LiNi0.5Mn1.5O4材料表面有約為2nm厚的類Mn3O4結構的薄層,與四面體的鋰位被過渡金屬離子部分占據有關。亞表層區域出現了巖鹽結構,由八面體空位被過渡金屬離子部分占據而引起,可以推斷出的是充電后的LiNi0.5Mn1.5O4材料中過渡金屬離子占據鋰位會阻礙Li+的擴散,導致電荷轉移阻抗增加,從而對LiNi0.5Mn1.5O4材料容量衰減以及首圈庫倫效率產生一定的影響。研究針對LiNi0.5Mn1.5O4材料中表面化學與電化學性能的關系從原子層面出發給出了基本的認知并拓寬了研究領域,就如何穩定鎳錳酸鋰材料的結構和提高其電化學性能的問題給出了建議,即將尖晶石LiNi0.5Mn1.5O4表面的四面體Li位被少量不溶性離子預先占據。當然,為了進一步理解充放電過程中表面結構的轉變對LiNi0.5Mn1.5O4電化學性能的影響,未來可以通過同時分析界面阻抗的變化、Mn離子的溶解、電極/電解液界面的穩定性來實現。
2.2 LNMO/電解液界面化學
2.2.1 電解液分解
鎳錳酸鋰材料充電到5 V高電位下,電解液易受材料中高氧化態過渡金屬離子Ni4+的誘導而發生分解,并且溶劑和電解質都會參與反應,反應產物沉積在正極/電解液界面層形成表面膜(簡稱CEI膜)。傳統碳酸酯類電解液由于LiPF6的存在,會發生分解形成PF5與LiF,由于微量水的存在,PF5又會轉變為LixPOyFz,而溶劑EC與DMC會被氧化形成聚醚與聚碳酸酯。因此,CEI膜主要由聚碳酸酯、聚醚等有機組分及LiF、LixPOyFz等無機組分構成。研究發現,電解液的降解產物形成的這層CEI膜并不能均勻包覆在正極材料表面,因此不能有效地保護LiNi0.5Mn1.5O4正極。而且,隨著循環的進行,電解液持續被反應,LiNi0.5Mn1.5O4正極上的表面膜不斷增厚,往往會導致電池阻抗持續增加及容量的衰減。
為了進一步改善LiNi0.5Mn1.5O4的循環性能,越來越多的研究人員開始關注高電壓電解液。一方面,開發耐5 V高電壓電解液,希望大大突破商業化電解液4.3 V的瓶頸;另一方面,開發新型添加劑,在正極表面更好地參與成膜反應,防止電解液與正極材料的直接接觸,進一步抑制電解液分解。在新型電解液體系方面,張文林等[22]合成了功能化離子液體1-丁基-3-甲基咪唑雙(三氟甲磺酰)亞胺鹽(BMIMTFSI)作為高壓鋰離子電池電解液組分,用于抑制有機溶劑的氧化,以提高碳酸酯類電解液的耐高壓性。結果表明,當在電解液中添加20%(體積分數)BMIMTFSI時,LiNi0.5Mn1.5O4/Li電池在室溫0.2C下的最高放電比容量是126.81 mA·h/g,5C下的放電比容量為109.36 mA·h/g,比在1 mol/L LiPF6-EC/DMC電解液中的放電比容量提高了91.7%,且該電池在0.2C下循環50圈后的放電比容量保持率在95%左右,比用碳酸酯類電解液提高了近10%。在新型添加劑方面,XU等[23]在碳酸酯基電解液中分別加入GBL(γ-丁內酯)和GLN(戊二腈)作為電解液添加劑,優先于溶劑分子發生氧化/還原分解反應,提高了電解液的氧化分解電壓,并在電極表面形成一層有效的保護膜,可抑制碳酸酯基溶劑的后續分解,提高電極/電解液界面穩定性,整體優化電池性能。
2.2.2 過渡金屬離子的溶解
金屬離子溶解是導致材料容量衰減的另一原因。 當LiNi0.5Mn1.5O4在高壓下工作時,由于電極 表 面 的 歧 化 反 應[24](2Mn3+→Mn2++Mn4+)以 及 電極和電解液之間的催化分解反應[25-27](由于微量水的存在,電解液中含有少量的HF,分解反應為:2Li Ni0.5Mn1.5O4+4H++4F-→3Ni0.25Mn0.75O2+0.75MnF2+0.25NiF2+2LiF+2H2O),導致過渡金屬離子的溶解。PIECZONKA等[25]研 究 了Mn和Ni在 不 同 條 件(如荷電狀態、溫度、存儲時間)下的溶解行為,發現LiNi0.5Mn1.5O4中Mn和Ni的溶解量隨著SOC(剩余電量)、溫度和儲存時間的增加而增加。這是因為乙醚是碳酸二乙酯(DEC)在電解液中的分解產物,隨著SOC、溫度和貯存時間的增加而增加。在乙醚的形成過程中(通過DEC的另一種分解產物乙醇的脫水反應)會產生額外的氫氟酸,加速了LNMO中錳和鎳的溶解。最近,JARRY等[27]提出了雙質子耦合電子轉移(PCET)反應機制用來描述LiNi0.5Mn1.5O4和碳酸酯電解液之間的界面反應過程,包括碳酸酯的電化學氧化及電極/有機電解液界面上錳和鎳的溶出,通過X射線、熒光光譜及成像實驗等表征手段證明碳酸酯在大于4.2 V的電壓下會發生電化學氧化,導致Ni2+、Mn2+/3+與β-二酮配體結合及Ni2+、Mn2+與碳酸根配體結合形成各種金屬絡合物,從而引起Ni/Mn的溶解,且隨著氧空位的增加,這種絡合反應的速率也會加快。要想減少這種界面反應的發生,降低過渡金屬的溶解,目前也有一些解決策略,比如可以優化正極材料的形貌、顆粒大小、晶面結構、包覆和摻雜等,電解液方面則可以通過優化溶劑、添加劑、離子液體等方式開發耐高壓的電解液。
3 LiNi0.5Mn1.5O4正極材料的改性研究
大量研究表明,通過表面包覆和離子摻雜可以有效解決上述表面及界面化學帶來的問題,極大提高電極材料的電化學性能。
3.1 表面包覆
考慮到電解液在高溫下更容易分解,并且分解產物并不能均勻包覆在正極材料表面,因此不能有效地保護LiNi0.5Mn1.5O4正極。而且隨著循環的進行,電解液持續被反應,LiNi0.5Mn1.5O4正極上的表面膜不斷增厚,生成的副產物還會加速Mn的溶解,導致電池 阻 抗 持 續 增 加 及 容 量 的 衰 減。PANG等[28]對LiNi0.5Mn1.5O4分別進行了SiO2和聚酰亞胺包覆,如圖5所示。由圖5研究表明,包覆對有序的LiNi0.5Mn1.5O4性能提升比無序的更明顯。除此之外,SiO2包覆后的LiNi0.5Mn1.5O4材料與未包覆的材料在25℃和55℃下的性能相比,容量衰減方面分別減少了45%和65%,較包覆之前容量衰減率明顯降低,循環性能大大提升;聚酰亞胺包覆的LiNi0.5Mn1.5O4材料在常溫下對性能的提升并不明顯,但高溫(55℃)下較包覆之前具有更低的容量衰減率,循環性能甚至優于SiO2包覆的LNMO材料,說明聚酰亞胺包覆層對材料的高溫穩定性更有利,這與聚酰亞胺優異的機械性能和化學穩定性相關。總之,包覆可以有效隔離電極和電解液,顯著改善材料的循環性能,選擇不同的包覆物對性能的提升程度至關重要。除了上述提到的氧化物、高分子聚合物外,目前包覆比較多的還有磷酸鹽[29-30],氟化物[31-32]等,雖然一定程度上改善了LiNi0.5Mn1.5O4材料的循環性能,但這類包覆層往往是電子與離子的不良導體,可能會對材料的倍率性能造成一定的負面影響,因此更多的研究者在努力尋找更為合適的包覆材料。

圖5 原始LNMO的FE-SEM圖(a1,a2)、SiO2-包覆LNMO的FE-SEM圖(b1)和TEM圖(b2)、PI-包覆LNMO的FE-SEM圖(c1,c2)Fig.5 FE-SEM images(a1,a2)of original LNMO,FE-SEM image(b1)and TEM image(b2)of LNMO coated with SiO2、and FE-SEM images(c1,c2)of LNMO coated with PI
3.2 離子摻雜
離子摻雜可以有效改善LiNi0.5Mn1.5O4正極材料的性能,目前研究最多的是包括Al[33-34]、Mg[35]、Fe[36-37]、Zn[38]、Cu[39]、Co[40-41]、Cr[42]等金屬離子在內的陽離子摻雜,通常金屬陽離子取代部分Ni或部分Mn,有以下幾點作用:1)抑制LixN1-xO等雜相生成;2)增大晶胞參數,有利于離子和電子傳輸;3)可誘導材料結構發生轉變(由P4332型轉變為性能更優的Fd3m型)。最近,LEE等[20]的研究表明,選用Cr-,Fe-和Ga-取代Ni-制備LiNi0.5-xMn1.5MxO4(M=Cr,Fe,Ga)材料,由于這幾種摻雜離子具有一定的自偏析效應,與Ni相比,更容易富集在材料表面而造成表面貧Ni的狀態,進而有效抑制電解液的氧化分解,從而改善材料的循環性能。除了陽離子摻雜外,目前也有一些陰離子摻雜來改善LiNi0.5Mn1.5O4性能的報道,多以F或S等陰離子取代部分O。以F元素摻雜為例,OH等[43]采用噴霧干燥法,以LiF為F源,制備的LiNi0.5Mn1.5O4-xFx(0 圖6 Cr-Al-F摻雜前后的LiNi0.5Mn1.5O4材料分別在1C下的循環性能Fig.6 Cycle performance of LiNi0.5Mn1.5O4 materials before and after doping with Cr-Al-F at 1C 尖晶石型鋰離子電池正極材料LiNi0.5Mn1.5O4具有高工作電壓平臺、高比容量、高能量密度、環境友好、循環性能好、三維鋰離子擴散通道等優點,在未來電動汽車、化學電池儲能等領域具有巨大潛在應用價值。但是,高工作電壓帶來的表面/界面等化學問題,大大制約了該材料的實際應用。本文從LiNi0.5Mn1.5O4正極材料的結構及脫嵌機理入手,對近年來LiNi0.5Mn1.5O4材料的研究進行了分析和總結,著重探討了表面/界面化學對LiNi0.5Mn1.5O4正極材料電化學性能的影響情況,旨在對LiNi0.5Mn1.5O4正極材料表面改性和電解液界面構筑方面提出一些思路。 1)電解液分解是目前制約LiNi0.5Mn1.5O4應用的最主要瓶頸,尋找耐高壓的電解液是關鍵,考慮到電解液與正極材料的兼容性,溶劑和添加劑的選擇未來還需要不斷優化。 2)正極材料的表面處理也起著重要的作用,離子摻雜與表面包覆可顯著改善材料的電化學性能,未來需要尋找更合適的摻雜離子,更優的包覆物,或者將摻雜和包覆結合起來,取長補短,利用兩者的協同效應,更有效地提高材料的電化學性能。 3)包覆方法上,也可以利用電解液添加劑在電極表面的優先分解對正極材料實現原位包覆,對包覆層的厚度及主要成分需要進一步研究。當然,也可以在正極材料表面構造濃度梯度,或者摻雜具有自偏析效應的金屬離子,使正極材料表面處于貧鎳狀態,盡可能地減少與電解液的副反應,或者采用不同的合成方法,控制晶面取向向電化學性能有利的方向生長等。 未來希望在提高LiNi0.5Mn1.5O4正極材料電化學性能的同時,能夠進一步探討其工作機理,為人們深入研究和開發LiNi0.5Mn1.5O4正極材料提供一定的指導依據,這些將有利于加速高比能量鋰離子電池正極材料的商業化進程。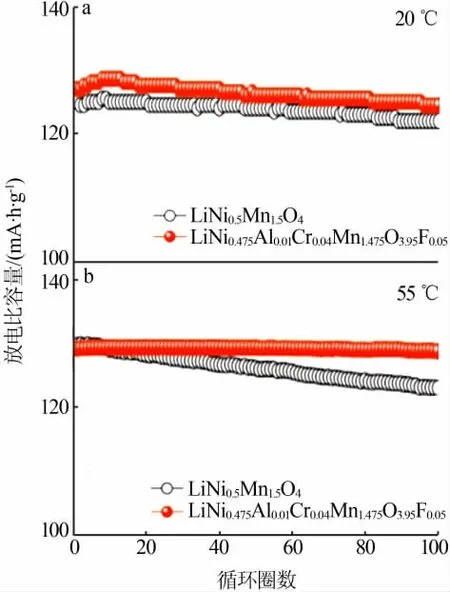
4 總結與展望
猜你喜歡
哲學評論(2021年2期)2021-08-22 01:53:34
當代陜西(2020年13期)2020-08-24 08:22:02
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
制造技術與機床(2017年5期)2018-01-19 02:49:17
金秋(2017年4期)2017-06-07 08:22:16
中國材料進展(2016年10期)2016-12-26 06:50:20
濰坊學院學報(2016年2期)2016-12-01 13:00:11
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
新聞傳播(2015年11期)2015-07-18 11:15:04
