黑曲霉GOD基因在枯草芽孢桿菌中的表達
2021-11-15 02:57:44于思穎張靜博曹平華李元曉馬彥博
養殖與飼料 2021年11期
于思穎 程 鵬 張靜博 曹平華 李元曉 馬彥博 李 旺
河南科技大學動物科技學院,河南洛陽 471023
葡萄糖氧化酶(glucose oxidase,GOD)是一類含有黃素腺嘌呤二核苷酸(flavine adenine dinucleotide,FAD)的二聚體蛋白酶,能夠特異性催化β-D-葡萄糖被氧化成葡萄糖酸和過氧化氫[1]。GOD 基于其安全無毒等特性廣泛地應用于飼料添加劑、食品、制藥和化學等領域[2-7]。
枯草芽孢桿菌(B.subtilis)作為一種革蘭氏陽性菌,已被美國食品藥品監督管理局(FDA)評為生物安全(Generally regarded as safe,GRAS)菌株[8],具有易于培養、繁殖速度快、遺傳背景清晰等特點。與大腸桿菌相比,其具有優異的蛋白分泌能力,方便下游表達產物的分離;同時,B.subtilis具有良好的發酵工藝技術,這為目的產物進一步擴大生產提供了基礎。B.subtilis表達系統已表達了多種產物并廣泛應用于制藥、食品和飼料等行業中[9-13]。
目前,大部分GOD 為野生菌或誘導菌生產,存在酶活力低且后續純化困難等問題,基因工程表達的GOD宿主多為畢赤酵母,由于畢赤酵母的表達往往需要甲醇誘導,難以直接應用于飼料和養殖生產。因此,本研究克隆了來源于黑曲霉的GOD 基因,以野生型動物益生菌為宿主,以期獲得高效表達GOD 的枯草芽孢桿菌,推動其在動物養殖中的應用。
1 材料與方法
1.1 試驗材料
1)土樣:采自河南省洛陽市某葡萄園,采用多點混合法采集0~10 cm的土壤。
2)菌株、質粒和引物見表1。

表1 本試驗所用菌株、質粒和引物
3)主要試劑:鄰聯茴香胺、辣根過氧化物酶、真菌DNA提取試劑盒購自上海生工生物有限公司;Taq DNA 聚合酶、dNTPs、DNA Marker、蛋白質Marker 購自Takara 公司;T4DNA 連接酶、NotI、NdeI、HindⅢ限制性內切酶購自Sigama 公司;質粒小提試劑盒、膠回收試劑盒購自天根生化科技(北京)有限公司;其他化學試劑均為國產分析純。
4)培養基。
①平板分離培養基:葡萄糖80 g/L、蛋白胨3 g/L、硫酸銨0.4 g/L、磷酸二氫鉀0.19 g/L、硫酸鎂0.16 g/L、碳酸鈣3.5 g/L、可溶性淀粉10 g/L、碘化鉀1.7 g/L、脫氧膽酸鈉0.2 g/L、加入適量鏈霉素(100 μg/mL)、瓊脂粉15 g/L、磷酸緩沖液0.1 mol/L,pH為5.6。
②發酵培養基:葡萄糖80 g/L、蛋白胨3 g/L、磷酸二氫鉀2 g/L、硫酸鎂0.7 g/L、氯化鉀0.5 g/L、硝酸鈉0.4 g/L、碳酸鈣3.5 g/L,自然pH。
③PDA 培養基:馬鈴薯200 g(加水1 L,煮沸30 min,紗布過濾),葡萄糖20 g,補水至1 L,自然pH,配制固體培養基時加入瓊脂粉20 g/L。
④LB 培養基:蛋白胨10 g/L,酵母膏5 g/L,NaCl 10 g/L;pH 7.0,121 ℃下滅菌20 min,配置固體培養基時加入瓊脂粉20 g/L。抗性培養基中Amp的最終質量濃度是100 μg/mL。
1.2 試驗方法
1)菌株篩選。采用Fiedure.K.J 顯色法,稱取10 g 采集的土樣置于滅菌的裝有100 mL生理鹽水且帶有適量玻璃珠的250 mL錐形瓶中,28 ℃,160 r/min振蕩培養30 min。然后用無菌水稀釋上清液至10-3、10-4、10-53個稀釋度,分別用無菌槍頭吸取100 μL 涂布于預先配制且滅菌的平板分離培養基,28 ℃培養3 d 后置于4 ℃冰箱中靜置,觀察藍紫色顏色圈,選取較大顏色圈中的菌株接種于PDA 斜面培養基,28 ℃培養至長出成熟孢子,保存于4 ℃。將初篩得到的斜面培養的菌株接種于發酵培養基中,28 ℃、180 r/min搖床振蕩培養5 d,取發酵上清液及菌體測酶活。
2)酶活力測定方法。
①葡萄糖氧化酶酶活的定義:在pH 5.6,溫度為30 ℃的條件下,每分鐘催化1 μmol葡萄糖轉化為葡萄糖酸和過氧化氫所需的葡萄糖氧化酶的量定義為1個酶活力單位(U)。
②底物體系:吸取1 mL 5%葡萄糖溶液,2 mL 0.07 g/L 鄰聯茴香胺溶液,0.1 mL 60 U/mg 辣根過氧化物酶溶液于同一試管中,30 ℃恒溫水浴10 min。
③酶活測定:吸取0.1 mL 稀釋10 倍后的粗酶液于底物中搖勻,于波長460 nm 處每1 min 記錄1次吸光度值(A460nm),共測定5 min,以A460nm對時間作圖,計算最大斜率ΔA(min),根據下式計算酶活(X):
X=ΔA*f/(11.3*t*V1/V2)
式中:f為粗酶液稀釋倍數;11.3 為消光系數;t為感應時間,min;V1為粗酶液體積,mL;V2為反應液總體積,mL。
3)基因組DNA的提取與目的基因的克隆。
①菌體的預處理:采用PDA 培養基平板活化待測菌株,于28 ℃培養至長出褐色孢子,之后接種于PDA 液體培養基,30 ℃、180 r/min 振蕩培養2~3 d,過濾出菌體,-80 ℃凍存。
②基因組DNA 的提取與鑒定:取凍存的菌體,在液氮中研磨,稱取0.2 g 研磨后的菌體,參照真菌DNA 抽提試劑盒提取待測菌株的基因組DNA,-20 ℃保存備用。采用真菌通用引物ITS1與ITS4 對5.8S rDNA-ITS 區序列進行PCR 擴增(94 ℃3 min,94 ℃30 s,55 ℃30 s,72 ℃2 min,運行35 個循環,72 ℃終延伸10 min),PCR 反應產物送至北京六合華大基因公司測序。
③目的基因的克隆:根據NCBI GenBank 上已公布的黑曲霉GOD 基因序列(登錄號:MH593586.1),設計克隆GOD 基因所用的特異性引物F、R。以黑曲霉基因組DNA 為模板,F、R 為引物進行PCR 擴增(98 ℃10 s,55 ℃30 s,72 ℃2 min,運行30 個循環),PCR 產物經純化回收后得到GOD 基因片段,連接pGEM-T Easy 載體,并轉化E.coliDH5α,通過藍白斑篩選,挑取白斑單菌落經菌落PCR 鑒定獲得陽性克隆子,提取pGEM-T-GOD 質粒雙酶切驗證后送測序,保存測序正確的菌株。
4)重組表達質粒的構建。結合枯草芽孢桿菌密碼子偏好性,對黑曲霉GOD編碼序列進行密碼子優化,優化后的氨基酸序列及核酸序列如下,在N端加上了34 個氨基酸信號肽(綠色部分),C 端加上了His-tag(紅色部分)。在核酸序列兩端添加NdeI(黃色序列)和HindⅢ(灰色序列)酶切位點,核酸序列全長1 902 bp,編碼629 個氨基酸,蛋白分子量約68.44 ku,序列交由公司(南京金斯瑞)合成。
氨基酸序列:MFAKRFKTSLLPLFAGFLLLFHLVLAGPAAASAE-Protein-HHHHHH..
核酸序列 pGOD:CATATGTTTGCAAAACGATTCAAAACCTCTTTAC
TGCCGTTATTCGCTGGATTTTTATTGCTGTTTC ATTTGGTTCTGGCAGGACCGGCGGCTGCGAGTGCTGAA-DNAsequence-CACCACCATCATCATCATTAATGAAAGCTT
分別酶切目的片段GOD 和載體質粒pT7M,用膠回收試劑盒將片段GOD 的酶切產物和質粒pT7M的酶切產物回收純化。在T4 DNA 連接酶的作用下構建重組質粒pT7M-GOD,轉化E.coli DH5α,經Amp(氨芐青霉素)抗性平板篩選和雙酶切鑒定,同時送至華大基因公司測序驗證,保存測序正確的菌株。
5)枯草芽孢桿菌的轉化及誘導表達。將鑒定正確的重組質粒pT7M-GOD通過化學轉化的方式轉入枯草芽孢桿菌7024E感受態中,在37 ℃、200 r/min搖床復蘇培養60 min 后涂于含有12.5 μg/mL 四環素的LB瓊脂平板上,37 ℃培養過夜。選取3株長勢良好的單菌落,分別接種于含有12.5 μg/mL 四環素的4 mL LB 培養基中,分為3 組,37 ℃、200 r/min 振蕩培養至OD600值達0.6~0.8 時,第1 組作為陰性對照,不添加誘導劑;第2組加入終濃度為2%木糖誘導表達,25 ℃誘導培養16 h;第3 組加入終濃度為2%木糖誘導表達,37 ℃誘導培養4 h。
6)目的基因的表達。收集培養物上清與菌體沉淀,在沉淀中加入裂解液(50 mmol/L Tris,150 mmol/L NaCl,5%甘油,pH 8.0),用超聲波破碎儀破碎。分別取培養物上清,全細胞裂解液,破碎液上清與沉淀,加入5×loading buffer 混勻,煮沸10 min,通過SDSPAGE 檢測粗蛋白的表達情況,通過制備單克隆抗體,利用Westen-blot檢測目的基因的表達。
2 結果與分析
2.1 產酶菌株的篩選與鑒定
通過對采集的土樣進行初篩和復篩,得到4 株產胞內GOD 酶活較高的菌株,測得酶活力如表2所示,由表2可以看出,菌株P-1 產酶性能較好,發酵5 d 后發酵液中GOD 酶活為2.820 U/mL,選定菌株P-1為出發菌株進行后續研究。
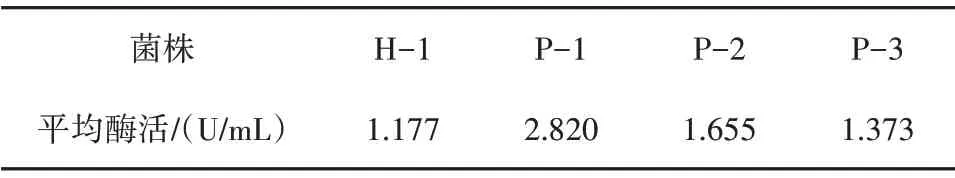
表2 分離菌株產GOD酶活測定結果
分析菌株P-1 的生長及形態特征,與曲霉鑒定手冊中黑曲霉比較接近。利用真菌通用引物ITS1/ITS4,擴增菌株P-1 的ITS 區基因,PCR 產物經瓊脂糖凝膠電泳,結果與理論編碼序列大小相符,如圖1所示。將測序得到的菌株P-1 IST 區基因序列在NCBI 中進行BLAST 分析,并構建系統發育樹見圖2。由圖2可知,與GenBank 數據庫中曲霉菌相關菌株的ITS 區基因序列的5.8S rDNA-ITS 區序列比較分析,發現其與曲霉屬(Aspergillussp.)的黑曲霉菌(Aspergillus niger)親緣關系最近。綜合菌株的菌落形態、生長特性及5.8S rDNA-ITS 區序列系統進化分析,確定菌株P-1為黑曲霉。
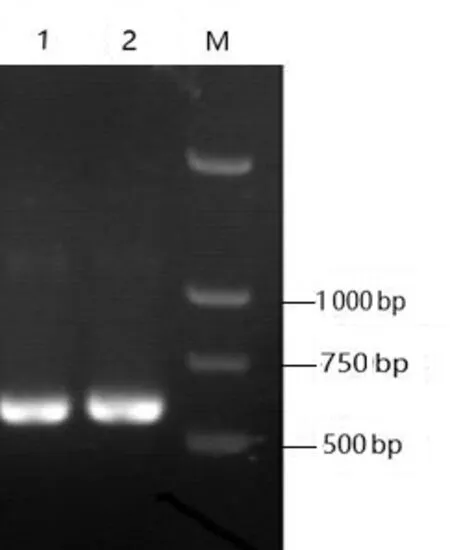
圖1 菌株P-1的IST區基因PCR產物
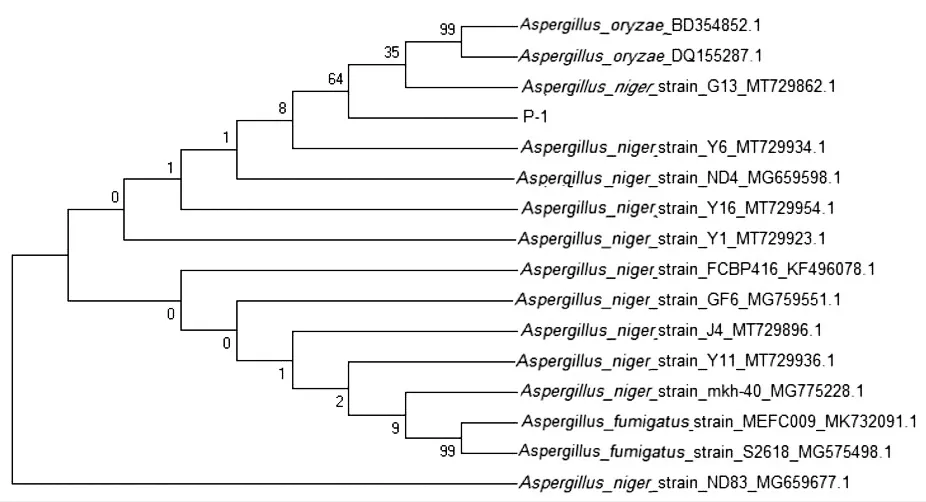
圖2 菌株P-1的5.8S rDNA ITS區序列系統發育樹
2.2 目的基因的克隆與鑒定
以黑曲霉P-1基因組DNA為模板,利用引物F、R 進行PCR 擴增,擴增產物經瓊脂糖凝膠電泳,結果如圖3A。質粒pGEM-T-GOD 經酶切,結果如圖3B。測序結果顯示,目的基因片段全長1 818 bp,編碼605 個氨基酸,將其序列與GenBank 中公布的GOD 基因序列進行比對,相似度為81%~99%,證明成功擴增了黑曲霉GOD基因,命名為pGOD。
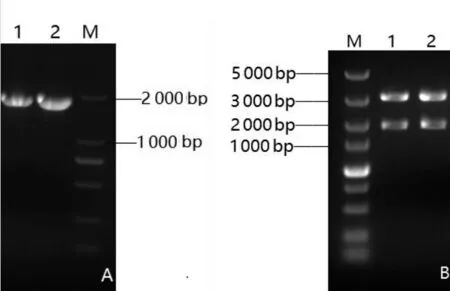
圖3 pGOD基因的PCR 擴增(A)和pGEM-T-pGOD的酶切鑒定(B)
2.3 重組表達質粒的構建與鑒定
重組質粒pT7M-GOD 經雙酶切驗證,結果顯示GOD 基因已經插入到表達載體pT7M 中(圖4)。重組質粒的測序結果顯示,目的基因序列與優化后pGOD 的序列完全一致,重組表達載體pT7M-pGOD構建正確。
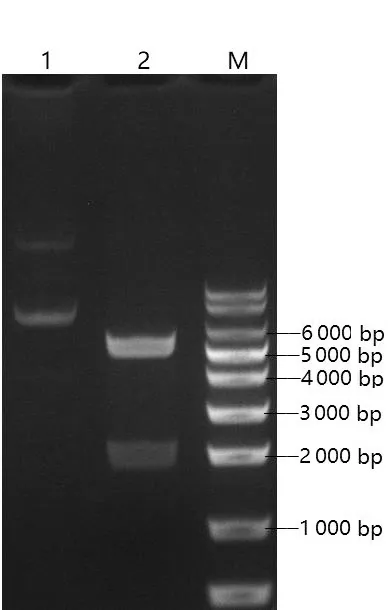
圖4 pT7M-GOD的雙酶切鑒定
2.4 目的基因的表達
將鑒定正確的重組質粒pT7M-pGOD 轉入枯草芽孢桿菌7024E 感受態中,經2%木糖誘導發酵培養后的SDS-PAGE 分析結果見圖5。結果顯示,1,2泳道和7,8泳道可見約68 ku大小的條帶,與理論上pGOD蛋白的分子質量大小一致。且2、8泳道明顯比1、7泳道的表達量高。結果表明,pGOD 基因能夠在枯草芽孢桿菌中表達,且在37 ℃培養4 h 表達量較高。
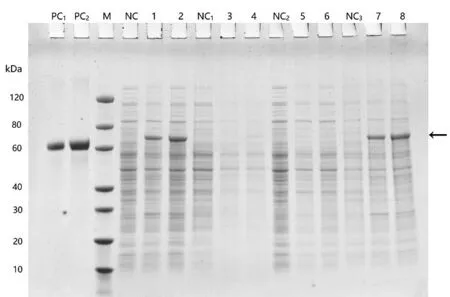
圖5 pGOD蛋白表達產物的SDS-PAGE
制備單克隆抗體,利用Westen-blot對表達的粗蛋白進行檢測,結果如圖6所示,在樣品3、4、5、6 中出現明顯的目的基因產物。大小與預期pGOD蛋白分子質量一致,說明該表達系統成功表達了pGOD基因,屬于胞內表達的可溶性蛋白。
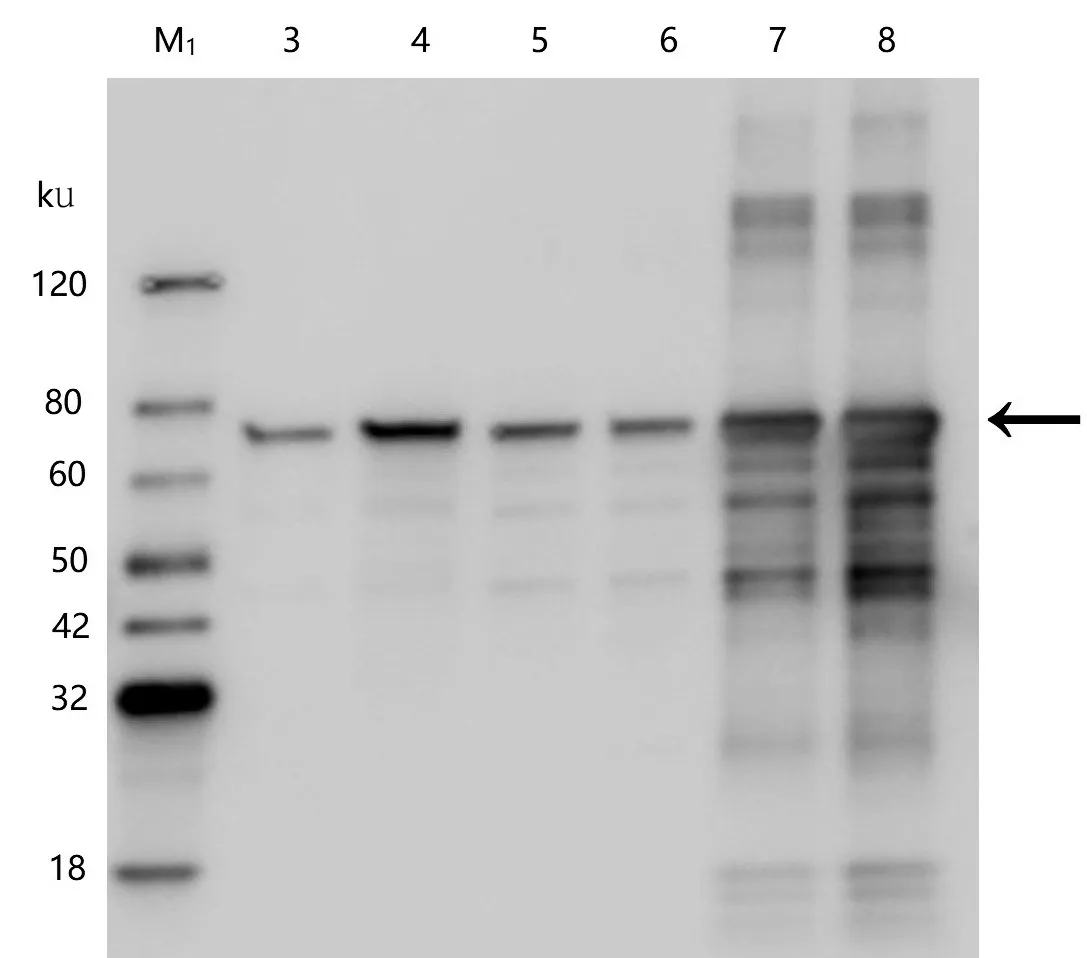
圖6 表達產物Western-blot檢驗
3 討 論
GOD 作為一種新型的酶制劑,近年來在養殖行業應用廣泛,GOD 可以改善動物腸道環境,增強腸道有益菌群,降低霉菌毒素中毒損傷,提高內源酶活,提高飼料消化率。大量的文獻報道表明產GOD的菌種主要是曲霉屬(Aspergillus)和青霉屬(Penicillium)[14-16],本研究從土壤樣品中篩選到產GOD 的微生物,經鑒定為黑曲霉,也驗證了這一結論,說明霉菌是產GOD的主要微生物菌種。
但天然菌株發酵生產GOD 的酶活性低,且純化工藝復雜。因此,構建高效表達GOD的重組菌株成為主要研究方向。近十幾年來,多種來源的GOD基因被克隆[17-18],本研究所擴增的GOD 基因片段全長1 818 bp,編碼605 個氨基酸,將其序列與GenBank中公布的GOD 基因序列進行比對,相似度為81%~99%,部分位點的堿基出現差異,其主要原因是不同物種的編碼基因有差異。所克隆的GOD 基因在大腸桿菌(Escherichia coli)、里氏木霉(Trichoderma reesei)、釀酒酵母(Saccharomyces cerevisiae)和畢赤酵母(P. pastoris)等宿主中成功表達[19-22],并且酶活性得到顯著提高。如陳楠等[23]利用具有AOX1 強啟動子的表達載體pPICZαA,在畢赤酵母SMD1168 中成功表達了黑曲霉PCTC GOD 基因,并對重組菌進行產酶條件優化,優化后酶活達32 U/mL,提高了27倍。然而在動物益生菌枯草芽孢桿菌中表達GOD基因的未見報道,B. subtilis屬于動物益生菌,發酵條件簡單,目前已有很多的研究報道使用B.subtilis作為高效生產的工程菌株[24-25]。加之為了更好地服務于養殖和飼料,本研究將GOD基因在野生型枯草芽孢桿菌中進行了表達,成功獲得了表達產物。但由于是異源表達,需對目的基因的密碼子進行優化[26-27],通過密碼子優化構建表達載體pT7M-pGOD,使枯草芽孢桿菌成功地表達了來源于黑曲霉的GOD基因,為構建高效表達GOD的基因工程菌提供了新的思路。
4 結 論
從土壤樣品中篩選出1 株高產葡萄糖氧化酶(GOD)的黑曲霉菌株,將黑曲霉菌的GOD基因克隆到載體pT7M,重組質粒pT7M-pGOD 轉化到枯草芽孢桿菌中異源表達,獲得了產GOD的枯草芽孢桿菌工程菌株。
