HPLC法同時檢測金櫻子果實中兩種三萜化合物的含量
2021-11-18 03:26:08高品一司星星張文超劉學貴李丹琦
沈陽化工大學學報 2021年3期
高品一, 司星星, 張文超, 劉學貴, 李丹琦
(1.沈陽化工大學 制藥與生物工程學院, 遼寧 沈陽 110142;2.沈陽化工大學 功能分子研究所, 遼寧 沈陽 110142;)3.沈陽化工大學 硼鎂資源開發與精細化工國家地方聯合工程實驗室, 遼寧 沈陽 110142;4.沈陽化工大學 遼寧省綠色功能分子設計與開發重點實驗室, 遼寧 沈陽 110142)
金櫻子(RosalaevigataMichx.)為薔薇科薔薇屬植物,又名刺頭、糖鶯子、糖橘子、棠球、燈籠果、藤勾子等,廣泛分布于安徽,兩湖等地[1-3].金櫻子果實成熟干燥后可入藥,是中國廣泛使用的傳統民間藥草,具有改善腎臟健康、固精縮尿、澀腸止帶、抑制動脈硬化和減少炎癥等作用[4-6].金櫻子果實中含有多種活性成分,包括三萜類、黃酮類、多糖類、鞣質類、酚酸類、甾體類等化合物,主要活性成分是三萜皂苷類,具有抗氧化、抗真菌和降糖降脂等作用[7].金櫻子的果實還可以用作健康食品和食品添加劑,如滋養口服液、果酒、醋和果醬等[8-10].目前關于金櫻子活性成分測定的研究報道多集中于總黃酮[11]、多糖[12]、總皂苷[13]、兒茶素[14]等.彭焱輝等用分光光度法測定了金櫻子果實中總皂苷的含量[13].本文對金櫻子果實中兩種三萜化合物皂苷及皂苷元的富集方法進行了研究.建立了HPLC法測定金櫻子果實中兩種三萜化合物含量的具體方法,為完善其質量標準和有效控制其質量提供依據.
1 實驗部分
1.1 儀器與材料
LC3000型高效液相色譜儀,北京創新通恒科技有限公司;promosil C18高效液相色譜柱,天津博納艾杰爾科技有限公司;R-3旋轉蒸發儀,瑞士步琪有限公司;電子天平BS-224S,賽多利斯科學儀器北京有限公司;SHZ-D(Ⅲ)循環水式真空泵,鞏義市予華儀器責任公司;KQ5200DE型數控超聲波清洗器,昆山市超聲儀器有限公司.
柱層層析硅膠(100~200、200~300、300~400目),青島海洋化工分廠;HSGF 254硅膠薄層層析硅膠板(20 cm×10 cm),青島海洋化工分廠;反向高效液相色譜柱,YMC-ODS-A-HG,北京慧得易科技有限責任公司;二氯甲烷,天津市大茂化學試劑廠;乙酸乙酯,石油醚,無水乙醇,天津市大茂化學試劑廠;其他試劑均為分析純,市售.
金櫻子藥材購買于沈陽市藥材市場,由沈陽藥科大學路金才教授鑒定為金櫻子RoseLaevigataMichx.的干燥果實;19α-羥基亞細亞酸(皂苷元)和19α-羥基亞細亞-28-O-β-D-吡喃葡萄糖苷(皂苷)標準品,沈陽化工大學化工與制藥研究室制備(樣本現保存在沈陽化工大學化工與制藥研究室).
1.2 有效成分的提取
分別稱取相同質量的金櫻子干燥果實粉末在相同條件下分別以微波法和超聲波法提取有效成分,處理后注入高效液相色譜儀進行分析.將金櫻子果實一部分去籽處理按超聲提取條件提取,另一部分不去籽提取,將提取液處理后注入高效液相色譜儀按實驗方法測定比較兩種化合物含量.
1.3 HPLC測定條件
色譜柱:promosil C18(4.6 × 250 mm,5 μm);檢測波長210 nm,柱溫15 ℃;流動相V(甲醇)∶V(水)=48∶52,流速0.9 mL/min.進樣量:25 μL,面積法計算含量.
1.4 樣品溶液的制備
標準樣品溶液制備流程:皂苷(19α-羥基亞細亞-28-O-β-D-吡喃葡萄糖苷)和皂苷元(19 α-羥基亞細亞酸)→減壓干燥→分別精密稱取5 mg,2.8 mg→無水甲醇溶解→10 mL容量瓶定容.
樣品溶液制備流程:(1)去籽金櫻子果實→干燥粉碎過40目篩→精密稱取5 g→V(二氯甲烷)∶V(甲醇)=5∶1定容于50 mL容量瓶→超聲處理30 min→減壓干燥→甲醇溶解定容于50 mL容量瓶→超聲處理30 min→甲醇定容;(2)未去籽金櫻子果實→干燥粉碎過40目篩→精密稱取5 g→V(二氯甲烷)∶V(甲醇)=5∶1定容于50 mL容量瓶→超聲處理30 min→減壓干燥→甲醇溶解定容于50 mL容量瓶→超聲處理30 min→甲醇定容.金櫻子果實樣品中有效成分的超聲提取條件為:超聲時間30 min、溫度35 ℃、超聲功率70 W.
2 結果與討論
2.1 金櫻子皂苷元的提取
采用冷凝回流提取工藝,以體積分數為85 %的乙醇水作為溶劑進行提取.14.5 kg干燥粉碎的金櫻子果實回流提取3次,流動相為石油醚,每次60 min;然后將金櫻子果實晾干,并將其用體積分數為85 %的乙醇水作為溶劑進行冷凝回流提取3次,每次2.5 h,并將粗提取液進行集中存放;使用旋轉蒸發儀進行減壓濃縮,濃縮至一定濃度使用電磁爐繼續揮發掉多余的乙醇,得金櫻子濃縮液,將濃縮液繼續揮發干溶劑變成浸膏;使用分液漏斗和鐵架臺對浸膏進行萃取;將適量的浸膏用蒸餾水溶解后轉移到分液漏斗中,加入等體積的有機溶劑二氯甲烷,進行反復萃取(約3次);萃取完成后得到有機層二氯甲烷和水層,在水層中加入等體積的乙酸乙酯,進行反復卒取3次;萃取完后得到乙酸乙酯層和水層,萃取后將二氯甲烷層、乙酸乙酯層和水層分別合并.
2.2 金櫻子皂苷元的純化富集
取萃取后的二氯甲烷層樣品與100~200目硅膠(拌樣質量比為1∶3)在研缽中充分拌樣;準備硅膠柱固體相,上樣,洗脫(流動相二氯甲烷和無水甲醇體積比分別為30∶1、20∶1、15∶1、10∶1、8∶1、6∶1、4∶1、2∶1,每組流動相沖洗5次);通過TLC檢測進行相同組分的合瓶.
將富集的減壓開放硅膠樣品以拌樣質量比為1∶3在研缽中充分拌樣;上樣,將拌好的樣品置于硅膠柱固體相上均勻平整分布,樣高約1.5 cm,然后將200~300目的硅膠裝入1 cm左右的玻璃柱中,放入適量棉花;以二氯甲烷和無水甲醇體積比為12∶1為流動相進行洗脫,洗脫到最后一瓶用TLC檢測無樣品為止;通過TLC檢測進行相同組分的合瓶.將富集到的樣品用適量無水甲醇進行溶解,然后上樣在ODS柱上,將其掛樣一夜,用體積分數50 %,55 %,60 %和70 %的甲醇水為流動相對其進行洗脫,每個梯度洗脫5次;通過TLC檢測進行相同組分的合瓶.經過ODS純化后得到金櫻子皂苷元純品1.5 g,HPLC(液相條件見1.3) 純度均大于95 %.
2.3 金櫻子皂苷元鑒定
將實驗得到的白色無定形粉末進行質譜和核磁譜分析.ESI- MS(m/z):527[M+Na]+.1H-NMR(600 MHz,C5D5N),δ:5.55(1H,br s,H-12),4.23(1H,dt,J=9.6,4.2 Hz,H-2),4.17(1H,d,J=10.8 Hz,H2-23),4.15(1H,d,J=10.8 Hz,H2-23),3.69(1H,d,J=9.6 Hz,H-3),3.06(1H,dt,J=8.4,2.4 Hz,H-16),3.01(1H,s,H-18),1.62(3H,s),1.48(3H,s),1.10(3H),1.09(3H,d,J=6.4 Hz),1.07(3H,s),1.04(3H,s).13C-NMR(150 MHz,C5D5N),δ:47.8(C-1),68.8(C-2),78.3(C-3),42.1(C-4),47.9(C-5),18.6(C-6),33.1(C-7),40.4(C-8),47.8(C-9),38.3(C-10),24.1(C-11),127.9(C-12),139.9(C-13),42.1(C-14),29.2(C-15),26.3(C-16),48.3(C-17),54.5(C-18),72.6(C-19),42.3(C-20),26.9(C-21),38.3(C-22),66.5(C-23),14.3(C-24),16.7(C-25),17.2(C-26),24.6(C-27),180.6(C-28),27.0(C-29),17.3(C-30).以上數據與文獻[15]基本一致,表明化合物是金櫻子皂苷元.
2.4 線性關系考察
精密吸取過濾后的標準樣品溶液5、10、15、20、25、30、40 μL,按給定色譜條件測定,以測得的峰面積為縱坐標,標準樣品進樣量為橫坐標進行線性回歸分析,結果如圖1所示.由圖1可以看出:皂苷標準曲線方程為y=5868.69x+6.655 8,R=0.997 0;皂苷元標準曲線方程為y=9945.21x-0.902 56,R=0.996 9.實驗結果表明皂苷和皂苷元含量在0.002~0.03 mg范圍內線性關系很好,適合作含量測定的標準曲線.
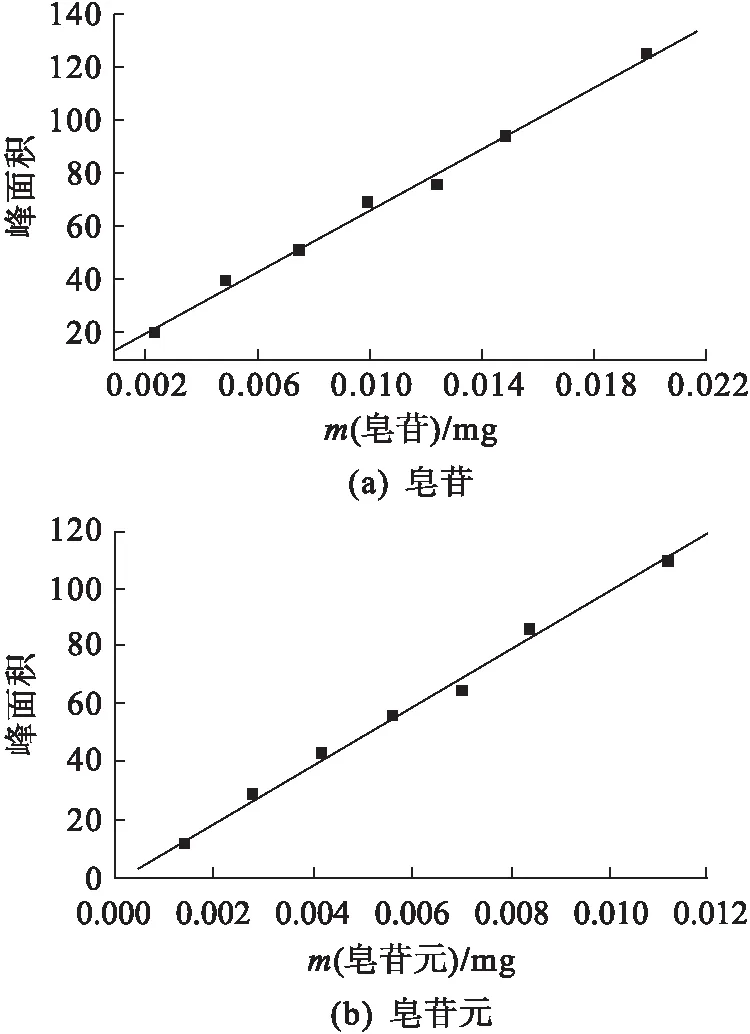
圖1 皂苷和皂苷元的標準曲線
2.5 精密度實驗
精密吸取經微孔濾膜過濾的標準樣品溶液10 μL,重復進樣5次,在1.3給定色譜條件下進行測定,記錄峰面積,測得皂苷和皂苷元的峰面積分別為39.169 3,29.118 7;39.457 9,29.688 1;39.604 7,29.552 9;39.682 5,29.616 8;39.145 4,29.109 4.皂苷峰面積的RSD為0.62 %,皂苷元峰面積的RSD為0.95 %,說明儀器系統及進樣方法精密度很好.
2.6 重現性實驗
精密稱取同一金櫻子干燥粉末3份,按樣品溶液制備流程制備樣品溶液,每份精密吸取25 μL,經微孔濾膜過濾的樣品溶液按照1.3給定的色譜條件進行測定,記錄峰面積.經計算總樣中皂苷和皂苷元峰面積的RSD分別為2.5 %和1.4 %,該結果說明此方法的重現性良好.
2.7 穩定性實驗
精密吸取同一份經微孔濾膜過濾的金櫻子樣品溶液25 μL,分別于0、3、6、9和12 h注入高效液相色譜儀進樣,按照1.3給定的色譜條件進行測定,記錄峰面積.比較所得到的5組峰面積,結果表明在12 h內進樣時,總樣中的皂苷及皂苷元等其他組分的檢測峰型穩定性很好.
2.8 加樣回收率的計算
取3份已測定2種化合物含量的去籽金櫻子干燥果實粉末各5 g,分別精密稱量3.2 mg、3.19 mg、3.2 mg皂苷和3.4 mg、3.39 mg、3.38 mg皂苷元標準樣品平行加入3份金櫻子中,按照上述標準樣品制備方法制備待測溶液,經微孔濾膜過濾后按1.3給定的色譜條件注入高效液相色譜儀中測定其峰面積,依據峰面積計算每份皂苷和皂苷元的含量,計算加樣回收率,結果如表1所示.

表1 金櫻子中皂苷和皂苷元的加樣回收率
由表1可以看出:皂苷和皂苷元的加樣回收率分別為101.56 %和102.94 %,此結果說明HPLC方法的重現性及準確度很好.
2.9 樣品含量測定
分別精密稱取3份去籽金櫻子干燥果實粉末、3份未去籽金櫻子干燥果實粉末各5 g.按樣品溶液制備方法制備樣品溶液,按1.3給定色譜條件測定峰面積,計算皂苷和皂苷元的含量結果見表2和表3.表2、表3結果表明:去籽金櫻子干燥果實中的皂苷和皂苷元含量高于未去籽金櫻子干燥果實,去籽后的平均含量皂苷為10.37 mg/g、皂苷元為5.36 mg/g,而未去籽金櫻子干燥果實的平均含量皂苷為8.39 mg/g、皂苷元為4.60 mg/g,建議提取兩類成分時將籽去掉以提高得率.

表2 去籽金櫻子果實中皂苷和皂苷元含量

表3 未去籽金櫻子果實中皂苷和皂苷元含量
3 結 論
隨著科技的不斷提高和創新,HPLC的應用也逐漸廣泛,并逐漸成為藥物分析的主要技術手段.HPLC法使用高效固定相,流動相采用高壓泵輸送,在線進行檢測,具有分離效能高、分析速度快、應用范圍廣、流出組分容易收集等優點,廣泛應用于藥物的含量測定.文章成功建立了同時測定金櫻子中皂苷(19α-羥基亞細亞-28-O-β-D-吡喃葡萄糖苷)和皂苷元(19α-羥基亞細亞酸)的高效液相色譜測定方法,面積歸一化法計算化合物含量,測得皂苷最多為10.37 mg/g、皂苷元為5.36 mg/g.提取金櫻子有效成分過程中考察了微波助提和超聲輔助提取兩種方法,文獻記載微波助提有很高的得率,但其選擇性強,對于同時多的提取兩種極性的成分結果不理想,因此以超聲法提取.為使總樣中的兩種成分盡可能多的被提取出來,并考慮兩種化合物的極性,先以二氯甲烷甲醇液提取,再以甲醇為溶劑提取,結果中總樣含量均很高.在提取過程中還考察了金櫻子果實去籽與未去籽時兩種成分的含量,結果表明去籽后皂苷及皂苷元的含量明顯高于去籽前的,說明兩種成分主要集中在金櫻子果皮中.該法可以用于金櫻子的質量控制.
