
湖南不同產地烏藥葉4種黃酮類成分的含量測定及其模式識別研究*
2021-11-21 13:08:16王瑞穎高元峰林麗美歐陽榮
中醫藥導報 2021年8期
易 柳,王瑞穎,高元峰,林麗美,歐陽榮
(1.湖南中醫藥大學第一附屬醫院,湖南 長沙 410007;2.湖南中醫藥大學藥學院,湖南 長沙 410207)
烏藥葉為樟科山胡椒屬植物烏藥Lindera aggregate(Sims)Kosterm的干燥葉,性辛、溫,歸脾、腎經,具有溫中理氣、消腫止痛的功效,主治脘腹冷痛、小便頻數、風濕痹痛、跌打傷痛、燙傷,其以葉片嫩綠時采摘質量佳[1]。烏藥葉藥用歷史悠久,在歷代古文獻中多有記載。烏藥葉最早收載于唐代《本草拾遺》:“炙研煎飲代茗,補中益氣,止小便滑數。”宋《開寶本草》也有記載:“烏藥……其葉及根嫩時采作茶片炙碾,服能補中益氣,偏止小便滑數。”明《食物本草》曰:“今人采……南燭、烏藥諸葉皆可為飲,以亂茶云。”明《本草蒙筌》載:“葉采入劑,下氣亦靈。但力遲緩,須醋浸炙。”明《本草綱目》曰:“……烏藥以產天臺者為勝,其嫩葉炙碾煎飲代茗,補中益氣,止小便滑數。”清《醫林纂要》云:“溫中燥脾,消食殺蛔。治腹中寒痛。”此外,張茂江等[2]對烏藥葉的臨床應用也有所報道,其常以烏藥葉外敷治癰疽、內服治胃痛。
現代研究表明,烏藥葉中含有氨基酸與蛋白質、有機酸、甾體皂苷、酚類與糅質、糖類、蒽醌、黃酮、香豆素與萜類內酯、強心苷等化學成分[3-5]。研究[6-11]發現烏藥葉主要含有倍半萜類和黃酮類化合物,其中黃酮類化合物是烏藥葉發揮藥理作用的主要活性物質。然而,現階段有關烏藥葉的研究并不多,且其質量標準未明,極大地限制了其臨床運用。故而,本研究采用HPLC同時測定烏藥葉中4個主要的黃酮類成分含量,旨在構建湖南省內不同地區烏藥葉的指紋圖譜并對其進行化學模式識別分析,以期為研究烏藥葉后續的藥效和作用機制提供物質基礎,為提高烏藥植物資源的整體利用率提供理論依據。
1 材料與儀器
1.1 材料與試劑 烏藥新鮮葉采自湖南省長沙市、湘潭市、益陽市、醴陵市、衡山縣等不同地區,共16批樣本,皆經湖南中醫藥大學第一附屬醫院歐陽榮教授鑒定為樟科山胡椒屬植物烏藥Lindera aggregate(Sims)Kosterm的葉。不同產地烏藥葉樣本的采集地和采集時間見表1。
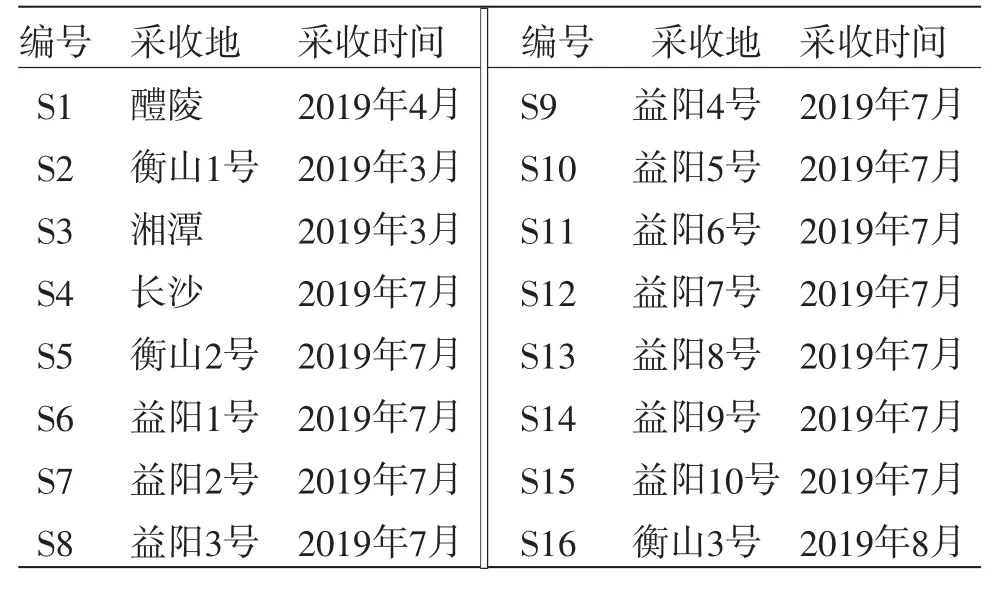
表1 烏藥新鮮葉采收地及采收時間
蘆丁對照品(批號:wkq19010203,純度:HPLC≥98%)、金絲桃苷對照品(批號:wkq19040913,純度:HPLC≥98%)、異槲皮苷對照品(批號:wkq19012403,純度:HPLC≥98%)、槲皮苷對照品(批號:wkq19031304,純度:HPLC≥98%)(四川省維克奇生物科技有限公司);甲醇、乙腈均為色譜純;磷酸為分析純;水為超純水。
1.2 主要儀器Agilent 1260高效液相色譜儀(美國安捷倫公司);Agilent HC-C18(2)(250 mm×4.6 mm,5 μm)色譜柱(美國安捷倫公司);DYQC-30型醫用超聲波清洗機(連云港歐倍潔醫療設備有限公司);WJX-A400型多功能高速搖擺粉碎機(上海緣沃工貿有限公司);AE2204電子分析天平(湖南湘儀天平儀器設備有限公司)。
2 方法與結果
2.1 色譜條件 色譜柱:Agilent HC-C18(2)(250 mm×4.6 mm,5 μm);流動相:乙腈(A)-0.4%磷酸溶液(B),梯度洗脫:0~3 min,12%A;3~26 min,12~22.5%A;26~42 min,22.5~24%A;42~45 min,24~12%A。流速:1.0 mL/min;柱溫:30℃;檢測波長:343 nm;進樣量:10 μL。
2.2 溶液制備
2.2.1 對照品溶液的制備 精密稱取真空干燥至恒重的對照品蘆丁0.005 8 g、金絲桃苷0.005 8 g、異槲皮苷0.007 2 g和槲皮苷0.005 6 g,分別置于10 mL容量瓶內,加入甲醇,定容,使其配成蘆丁0.58 mg/mL、金絲桃苷0.58 mg/mL、異槲皮苷0.72 mg/mL和槲皮苷0.56 mg/mL的單一對照品溶液。
2.2.2 供試品溶液的制備 將采摘的烏藥新鮮葉用清水洗凈,自然風干,115℃殺青20 min,70℃下干燥7 h,粉碎后過0.45 mm(40目)金屬網篩。
精密稱取上述干燥的烏藥葉粉末1.0 g,置錐形瓶中,精密加入甲醇50 mL,浸泡1 h,稱定質量,超聲處理1 h,放冷,加甲醇補足減失質量,搖勻,過濾,棄去初濾液,收集續濾液。將續濾液稀釋5倍,過0.22 μm微孔濾膜,即得供試品溶液。
2.3 方法學考察
2.3.1 專屬性考察 分別取空白溶劑、混合對照品溶液和供試品溶液,在上述色譜條件下依法測定。結果顯示,4種對照品色譜峰相應的保留時間處,空白溶劑無干擾,各成分峰形較好且均能達到基線分離,分離度均大于1.5;理論塔板數按槲皮苷計均不低于5 000。
2.3.2 線性關系考察 精密量取混合對照品溶液,將混合對照品溶液用甲醇稀釋成一系列的梯度濃度溶液后,按照相應的色譜條件測定。以待測化合物濃度對峰面積進行線性回歸,得到各成分標準曲線。結果顯示,在定量范圍內各化合物的線性良好,各成分回歸方程及相關系數見表2。
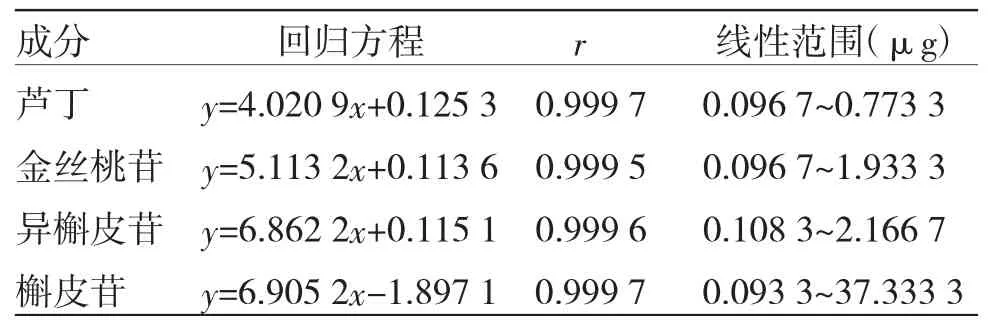
表2 各成分回歸方程及相關系數
2.3.3 精密度試驗 精密稱取烏藥葉樣品1.0 g,按上述方法制成供試品溶液,連續進樣6次,測得蘆丁、金絲桃苷、異槲皮苷和槲皮苷峰面積的RSD(n=6)分別為0.60%、1.60%、0.94%、0.46%,表明儀器精密度良好。
2.3.4 穩定性試驗 取同一份供試品溶液按照上述色譜條件分別在0、2、4、8、12、24 h進樣,測得蘆丁、金絲桃苷、異槲皮苷和槲皮苷峰面積的RSD(n=6)分別為0.12%、1.20%、1.57%、0.28%,表明供試品溶液在24 h內穩定性良好。
2.3.5 重復性試驗 精密稱取烏藥葉樣品6份,每份約1.0 g,按照上述方法平行制備6份供試品溶液,分別進樣,測定蘆丁、金絲桃苷、異槲皮苷和槲皮苷的峰面積,計算RSD值分別為1.82%、1.90%、1.62%、1.17%,表明重復性良好。
2.3.6 加樣回收率試驗 精密稱取醴陵烏藥葉樣品6份,每份約1.0 g,精密稱定,分別精密加入蘆丁、金絲桃苷、異槲皮苷、槲皮苷對照品溶液0.130、0.260、0.375、14.500 mL,再加入甲醇34.735 mL,按照上述方法制備供試品溶液,分別進樣,測定蘆丁、金絲桃苷、異槲皮苷和槲皮苷的峰面積,加樣回收率見表3。

表3 各成分的加樣回收率測定結果(n=6)
2.4 HPLC法同時測定烏藥葉中蘆丁、金絲桃苷、異槲皮苷、槲皮苷的含量 將16批烏藥葉樣本按照“2.2.2”項下的方法制備供試品溶液,并按照“2.1”項下的色譜條件,分別進樣,記錄蘆丁、金絲桃苷、異槲皮苷和槲皮苷的峰面積,通過回歸方程,計算各樣本中蘆丁、金絲桃苷、異槲皮苷和槲皮苷的含量。(見表4)
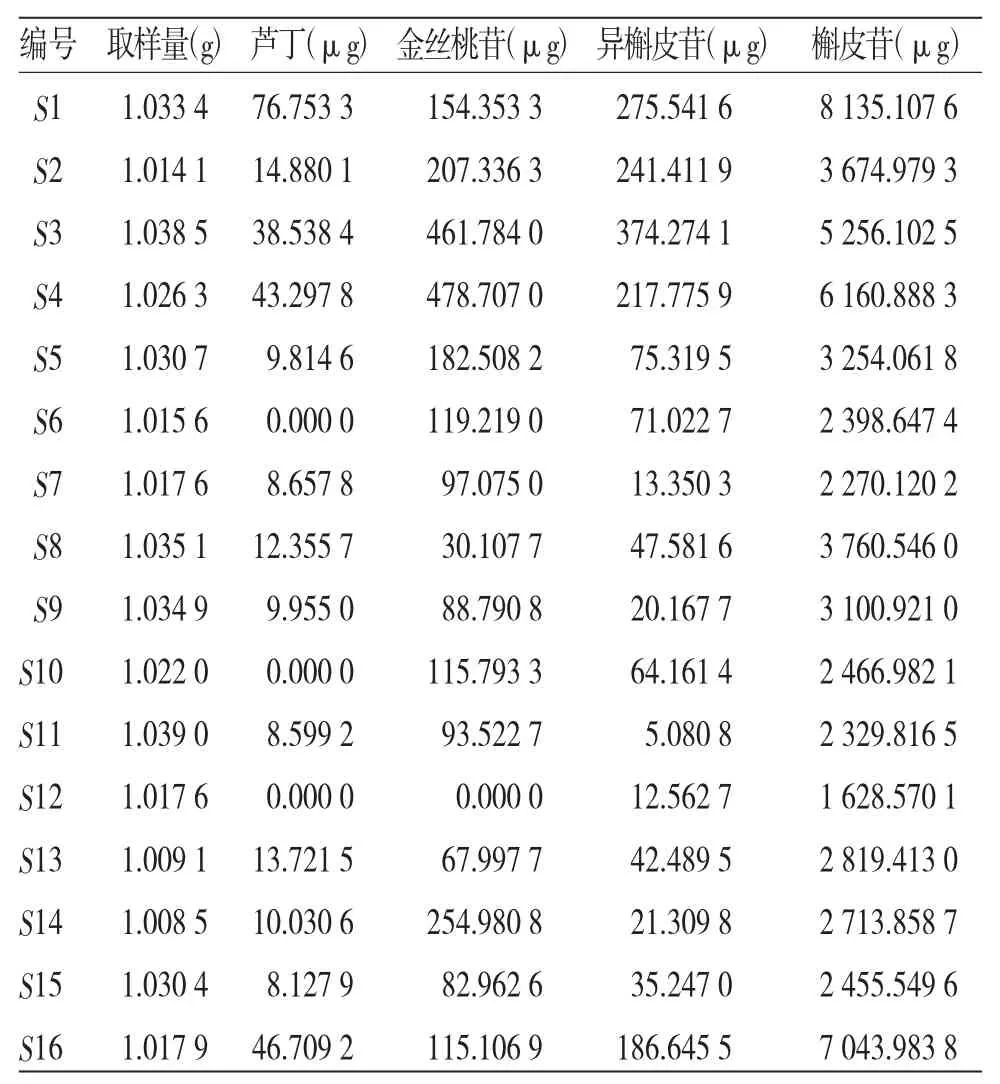
表4 不同產地烏藥葉藥材中黃酮類成分的含量測定(n=3)
2.5 指紋圖譜的建立及相似度分析
2.5.1 指紋圖譜的建立 取不同產地的16批烏藥葉,按“2.2.2”項下樣品處理方法制備供試品溶液,按“2.1”項下色譜條件進樣檢測,采用“2012版中藥色譜指紋圖譜相似度評價系統”,以S1為參照圖譜,時間窗寬度為0.1,通過多點校正、全譜峰匹配,得到16批烏藥葉指紋圖譜共有模式及對照譜圖譜,見圖1。經匹配后標定了9個共有峰,經過與對照品色譜圖及保留時間對比,發現異槲皮苷和槲皮苷峰最為明顯。
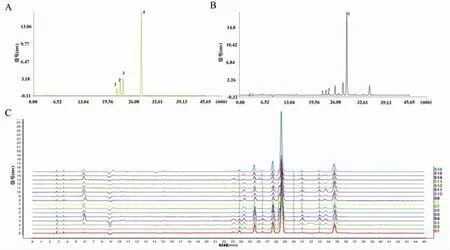
圖1 16批烏藥葉指紋圖譜圖
2.5.2 相似度評價 以對照圖譜為參照,對16批烏藥葉指紋圖譜進行相似度評價,結果16批烏藥葉相似度為0.840~0.989,表明相似度良好。(見表5)
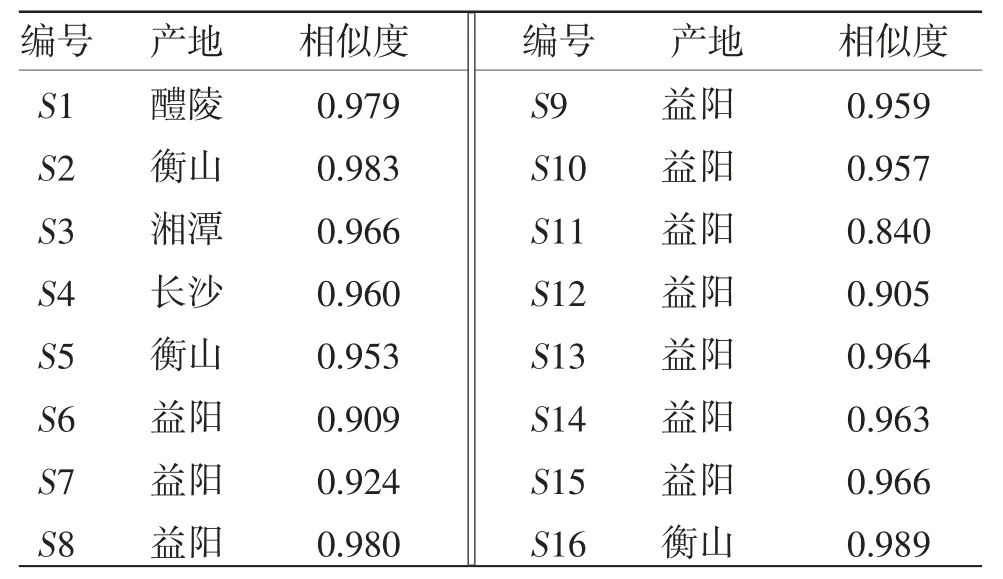
表5 不同產地烏藥葉生品相似度分析
2.6 化學模式識別分析
2.6.1 聚類分析 應用SPSS 24.0軟件,以共有峰的峰面積為變量,進行系統聚類分析。結果顯示,16批烏藥葉樣本可分為4類,Ⅰ類:S3~S4,Ⅱ類:S1、S16,Ⅲ類:S2、S5、S8,Ⅳ類:S6~S7、S9~S15。其中S6~S7、S9~S15均采自湖南益陽地區,由此說明同一產地不同批次樣本間差異較小。Ⅰ類中S3、S4分別采自湘潭、長沙;Ⅱ類中S1、S16分別采自醴陵、衡山;Ⅲ類中S2、S5采自衡山,S8采自益陽,Ⅰ類、Ⅱ類、Ⅲ類中的某些樣本雖采自不同地區,卻歸為同一類,推測可能與不同批次樣本的生長環境和采收時間等有關。(見圖2)
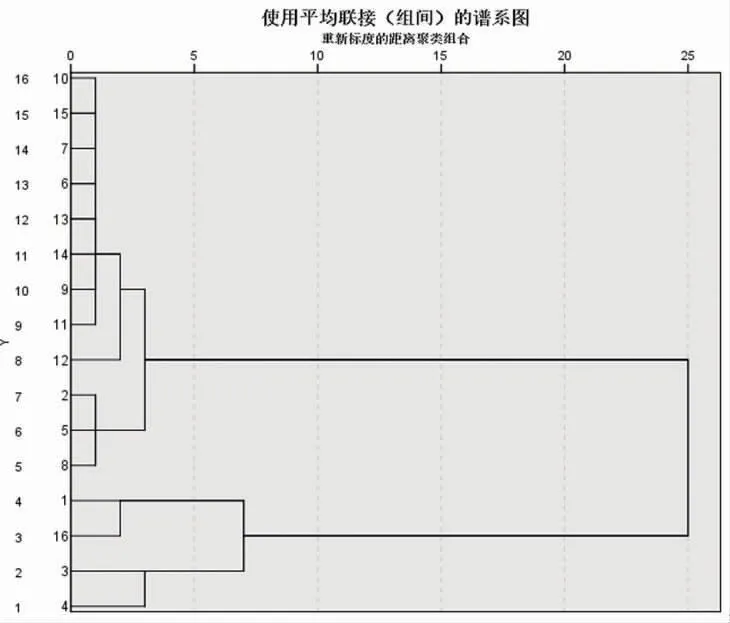
圖2 16批烏藥葉聚類分析
2.6.2 主成分分析 應用SPSS 24.0軟件對數據標準化后進行主成分分析,首先篩選出特征值大于1且貢獻率大于85%的成分,結果發現差異較大的2個主成分的特征值均大于1,其累計貢獻率大于85%,即這2個主成分包含了9個共有成分的89.002%的信息,符合主成分分析條件。結果烏藥葉第一主成分特征值為6.267,貢獻率為69.632%;第二主成分特征值為1.743,貢獻率為19.370%(見表6)。碎石圖結果顯示,上述的2個主成分的坡度較陡,表明這2個主成分是評價烏藥葉質量的代表性成分(見圖3A)。以9個共有峰的峰面積為變量,導入SIMCA 16.0軟件,繪制主成分得分圖(見圖3B)。結果表明主成分分類結果與聚類分析結果一致,但長沙地區的烏藥葉樣本不在置信區間內,結合兩項分析,說明同一產地不同批次樣品間差異較小,不同地區烏藥葉的質量仍存在著一定的差異。

表6 烏藥葉主成分特征值及累計方差貢獻率

圖3 16批烏藥葉主成分分析結果
3 討 論
湖南地區的烏藥植物資源十分豐富。目前,烏藥植物主要取其塊根入藥[12],而地上部分的藥用價值尚未被開發,出現“湖南烏藥作柴燒”的現象,以致烏藥植物資源嚴重浪費。盡管烏藥葉在歷代文獻中多有記載,藥用歷史悠久,但臨床上烏藥葉的藥用利用率極低。化學成分研究發現,烏藥葉中含有多類成分,其中黃酮類成分是其發揮藥理作用的主要活性物質,然而烏藥葉的質量標準不清。參考其他文獻[13-20],本研究構建了烏藥葉黃酮類成分含量測定方法。(1)提取方法考察方面:進行單因素考察,對比了甲醇超聲提取與70%乙醇回流提取的區別,發現兩種提取方法所得烏藥葉中的黃酮類成分含量差異不明顯,且超聲提取方便、快捷,故而本研究采用甲醇超聲法制備供試品溶液。(2)HPLC方法構建方面:①波長方面,考察了250~360 nm波段,發現在343 nm的條件下,基線較穩且峰形最好,最終確定343 nm作為檢測波長;②進樣量方面,考察了10、20 μL,根據實際需要以及峰形峰寬,確定進樣量為10 μL;③柱溫方面,考察了25~35℃,根據色譜圖峰形、峰高,確定柱溫為30℃;④流速方面,考察了1.0、1.2 mL/min,從色譜圖峰形、峰高來看,兩種流速無明顯差異,考慮到色譜柱的使用壽命,最終確定流速為1.0 mL/min。此外,在上述確定的HPLC條件下,我們對烏藥葉中的槲皮素和山奈酚也進行了含量考察分析,發現其含量極低,故在本研究中未采用這兩個成分作為定量分析的指標。本研究對烏藥葉中的4個黃酮類成分進行含量測定,結果顯示16批烏藥葉樣品中各成分的含量高低不同,說明不同地區烏藥葉的質量存在著一定差異。
本研究通過分析湖南多個產地16批烏藥葉指紋圖譜,共確認了9個共有峰,指認了其中2個重要成分,分別為異槲皮苷、槲皮苷。在色譜分析的基礎上,采用聚類分析對16批烏藥葉樣本進行研究,發現具有明顯的分類趨勢,大致分為4類,其中個別樣品與同產地其他批次樣品差異較大,這可能與樣品采收季節等條件有關。在不同生長時期,植物內各成分的含量隨時間變化,這提示著藥材需在適宜的時間采收,才能保證藥材的質量。我們采用主成分分析篩選出導致樣品不同分類差異的主成分,發現這2個主成分的累計方差貢獻率為89.002%,說明了前2個主成分能夠概括原有數據的絕大部分信息,推測異槲皮苷、槲皮苷對主成分有較大影響,為烏藥葉的多成分含量測定提供了依據。
