虎杖解毒顆粒(無糖型)改制前后指紋圖譜對比研究
2021-12-03 05:04:16白雨鑫李晉齊童榮生
實用藥物與臨床 2021年11期
白雨鑫,李晉齊,童榮生
0 引言
中藥指紋圖譜研究技術以復方制劑整體為研究對象,展示制劑品質的真實性、一致性和穩定性,全面評價制劑中復雜的化學成分和比例,已成為國際公認的中藥材和各制劑質量控制手段[1-2]。虎杖解毒顆粒(無糖型)是在保證原虎杖解毒顆粒處方、提取以及精制工藝不變的情況下,對成型工藝改制而得。為評價改制前后質量的變化,本研究對這兩種制劑分別建立指紋圖譜,并比較其共有峰及共有圖譜模式的差異。
1 試驗材料
1.1 藥物與試劑 虎杖解毒顆粒(無糖型)和虎杖解毒顆粒(四川省人民醫院藥學部制劑室,各10個不同批次)、大黃素(批號:110756-200110)、虎杖苷(批號:11575-200502)、射干苷(批號:111632-200501),甲醇、乙腈為色譜純,磷酸為分析純,試驗用水為超純水。
1.2 儀器 e2695型HPLC儀(包括Waters公司2998 PDA檢測器,Empower3色譜工作站);BP211D型電子分析天平(十萬分之一,德國Sartorius公司);KQ-500DB型數控超聲波清洗器(昆山市超聲儀器有限公司)。
2 方法與結果
2.1 色譜條件 色譜柱:Phenomenex Gemini 110A C18(250 mm×4.6 mm,5μm);檢測波長300 nm;柱溫30 ℃;進樣量10 μl;流動相:乙腈(A)-0.05%磷酸溶液(B),梯度洗脫(見表1);色譜記錄時間為120 min[3-4]。
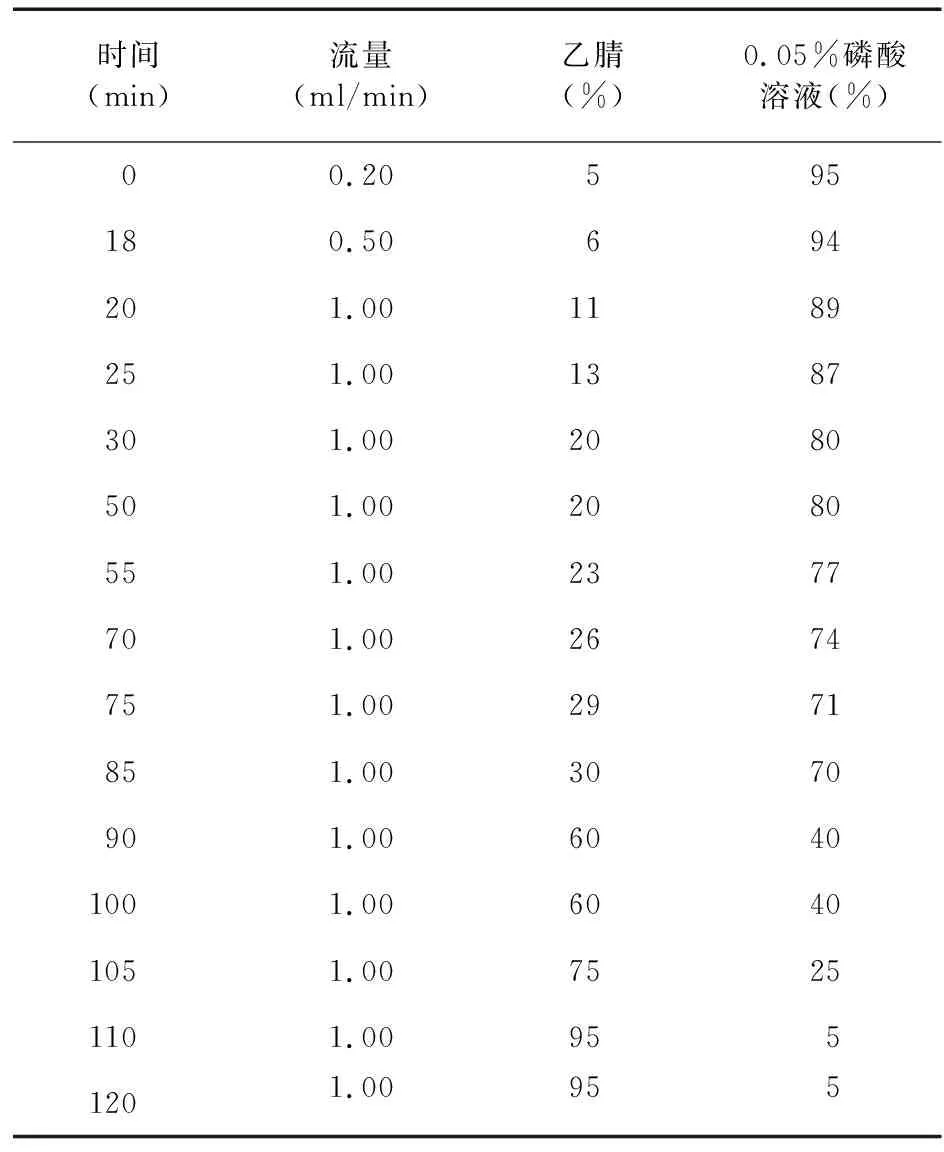
表1 流動相梯度表
2.2 溶液制備
2.2.1 混合對照品溶液 取虎杖苷、射干苷、大黃素對照品適量,各精密稱定后置于50 ml容量瓶中,甲醇超聲溶解,放冷定容,搖勻得虎杖苷8.83 μg/ml、射干苷15.15 μg/ml、大黃素7.28 μg/ml的對照品溶液。
2.2.2 供試品溶液 取研細并過三號篩的虎杖解毒顆粒(無糖型)粉末近5 mg,精密稱定,轉移至50 ml棕色容量瓶中,加入40 ml甲醇,超聲溶解60 min,放至室溫后定容搖勻,用0.45 μm有機系微孔濾膜濾過5 ml,取續濾液,即得。同法制備虎杖解毒顆粒供試品溶液。
2.3 方法學考察
2.3.1 重復性試驗 以編號S1的同一供試品為對象,按照“2.2.2”項下方法平行制備6份供試品溶液,按“2.1”項下色譜條件進樣10 μl測定。以7號峰(虎杖苷)為參照,計算出各共有指紋峰的相對保留時間RSD范圍(0.28%~0.62%)和相對峰面積RSD范圍(0.59%~2.13%),表明建立的方法重復性良好。
2.3.2 穩定性試驗 取制備的供試品(編號S1)溶液,按“2.1”項下色譜條件,分別于0、2、4、6、12、24 h進樣10 μl,以7號峰(虎杖苷)為參照,計算出各共有指紋峰的相對保留時間RSD范圍(0.27%~0.79%)和相對峰面積RSD范圍(0.46%~2.07%),表明該方法制得的溶液穩定性良好。
2.3.3 精密度試驗 取制備的供試品(編號S1)溶液,按“2.1”項下色譜條件連續進樣6次,每次10 μl,分析圖譜。以7號峰(虎杖苷)為參照,計算出各共有指紋峰的相對保留時間RSD范圍(0.16%~0.57%)和相對峰面積RSD范圍(0.37%~1.83%),表明儀器精密度良好。
2.4 指紋圖譜的建立與分析
2.4.1 指紋圖譜的重疊匹配 取10批虎杖解毒顆粒(無糖型)和虎杖解毒顆粒樣品,以“2.2.2”項下方法處理,“2.1”項下色譜條件測定。采用中藥色譜指紋圖譜相似度評價系統(2012版)導入數據,分析得到兩種制劑樣品的指紋圖譜重疊匹配圖[5],結果見圖1、圖2。
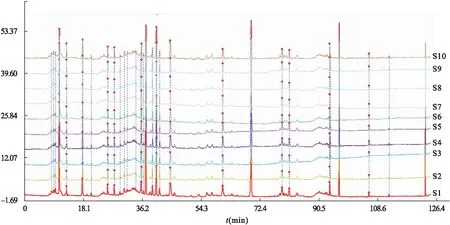
圖1 虎杖解毒顆粒(無糖型)的指紋重疊色譜圖

圖2 虎杖解毒顆粒的指紋重疊色譜圖
2.4.2 共有峰的標定和參照峰的確認 利用多點校正進行自動匹配,中位數法生成對照圖譜,共標定了19個峰形較好、穩定出現的共有峰。其中7號峰吸收強,峰面積大且保留時間穩定,故將其作為參照峰。
2.4.3 特征成分的指認 精密吸取虎杖解毒顆粒(無糖型)、虎杖解毒顆粒和混合對照品溶液,以“2.1”項下色譜條件進樣分析,色譜圖匹配后見圖3。通過與對照品圖譜比對,指認兩種供試品溶液的特征共有峰,分別為虎杖苷、射干苷和大黃素,7號參照峰為虎杖苷。
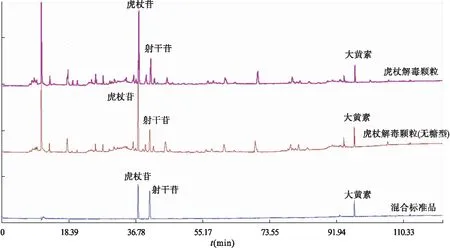
圖3 虎杖解毒顆粒、虎杖解毒顆粒(無糖型)和混合對照品匹配色譜
2.4.4 虎杖解毒顆粒(無糖型)和虎杖解毒顆粒的共有模式比較 利用中藥色譜指紋圖譜相似度評價系統(2012版),得到兩種供試品制劑的指紋圖譜共有模式,如圖4所示。
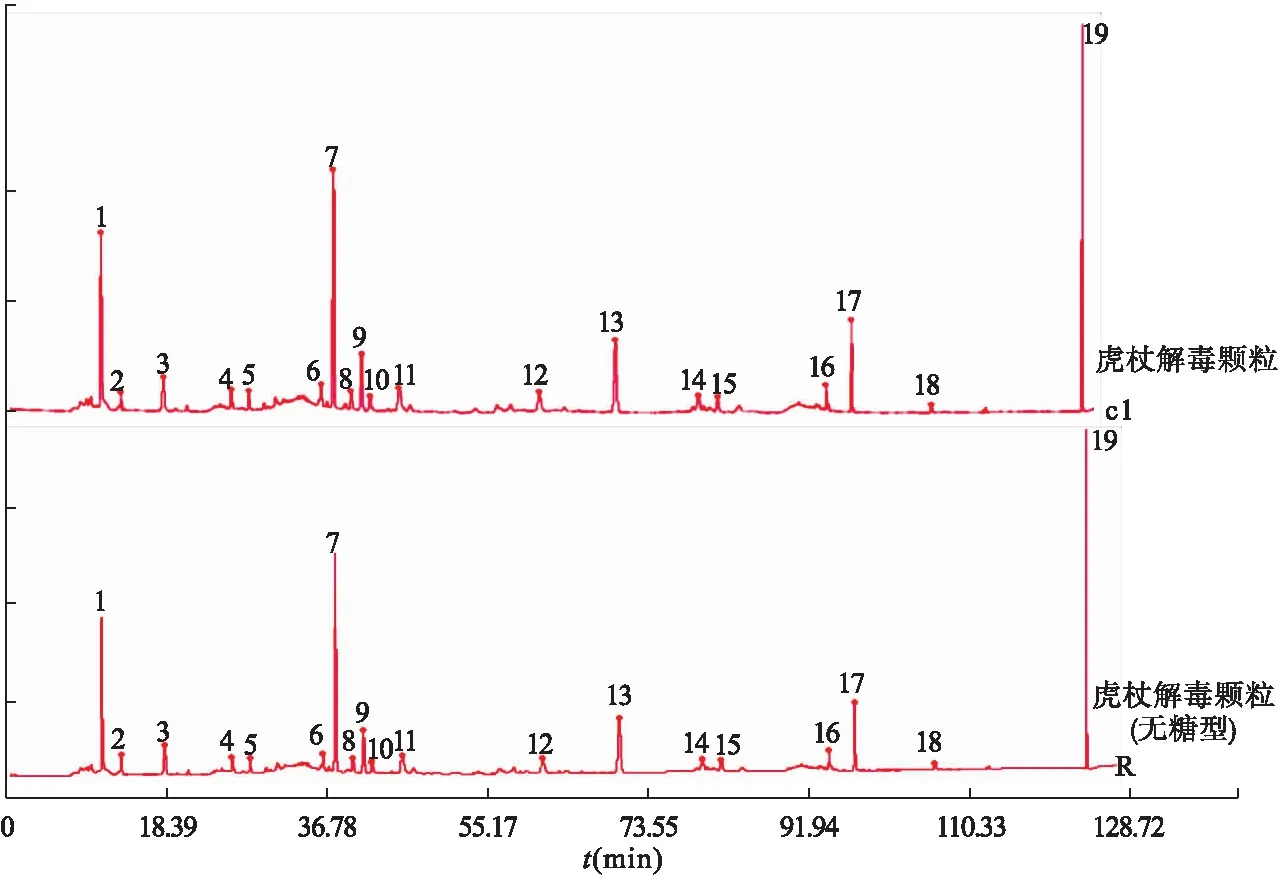
圖4 虎杖解毒顆粒和虎杖解毒顆粒(無糖型)共有模式注:7.虎杖苷;9.射干苷;17.大黃素
2.5 指紋圖譜的相似度分析
2.5.1 指紋圖譜中共有峰的相對保留時間和相對峰面積 以參照峰(峰7)的保留時間和色譜峰面積為1.000 0,分別計算虎杖解毒顆粒(無糖型)和虎杖解毒顆粒中共有峰(1~19號峰)的相對保留時間和相對峰面積,結果見表2~表5。
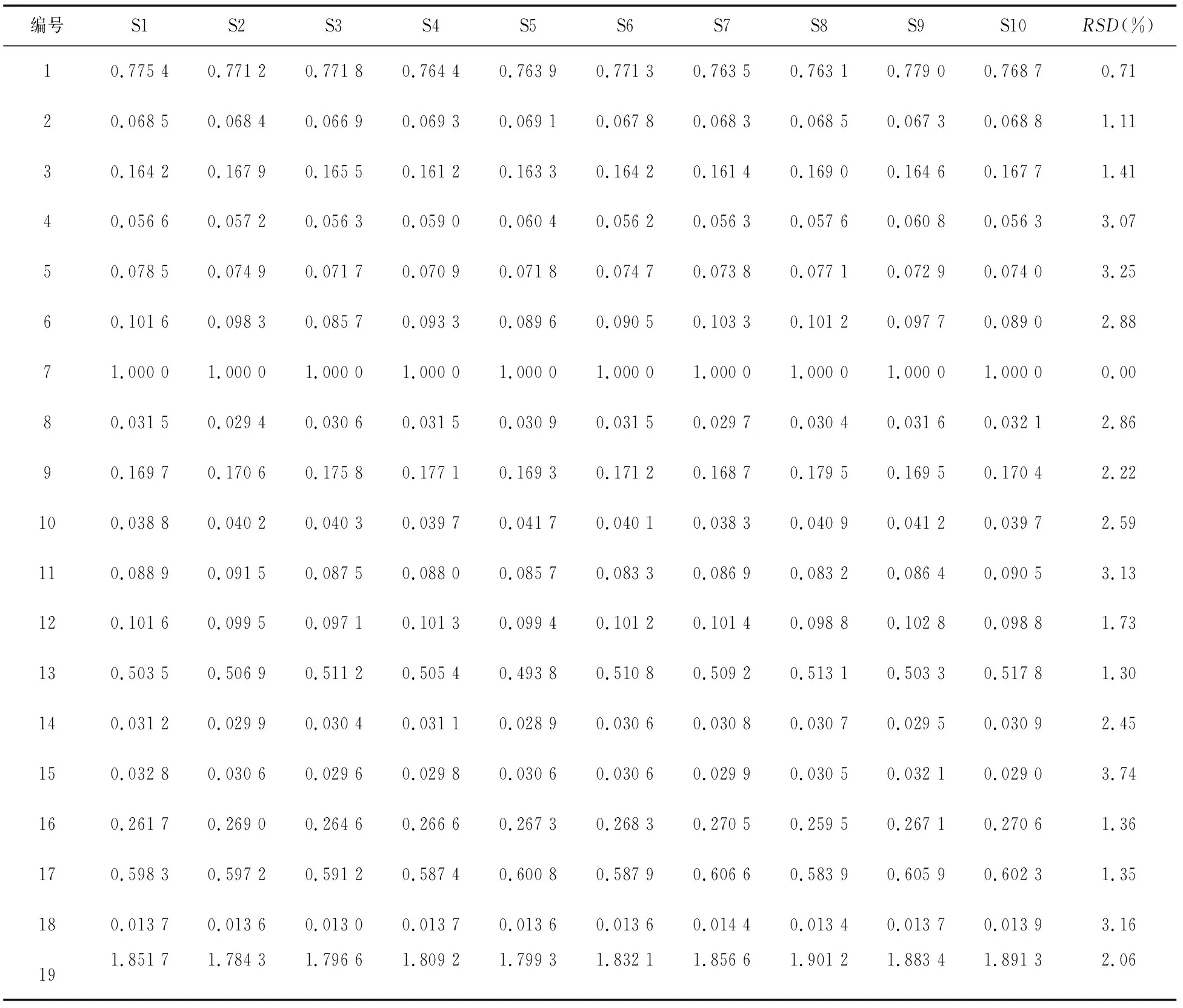
表2 虎杖解毒顆粒(無糖型)共有峰的相對峰面積
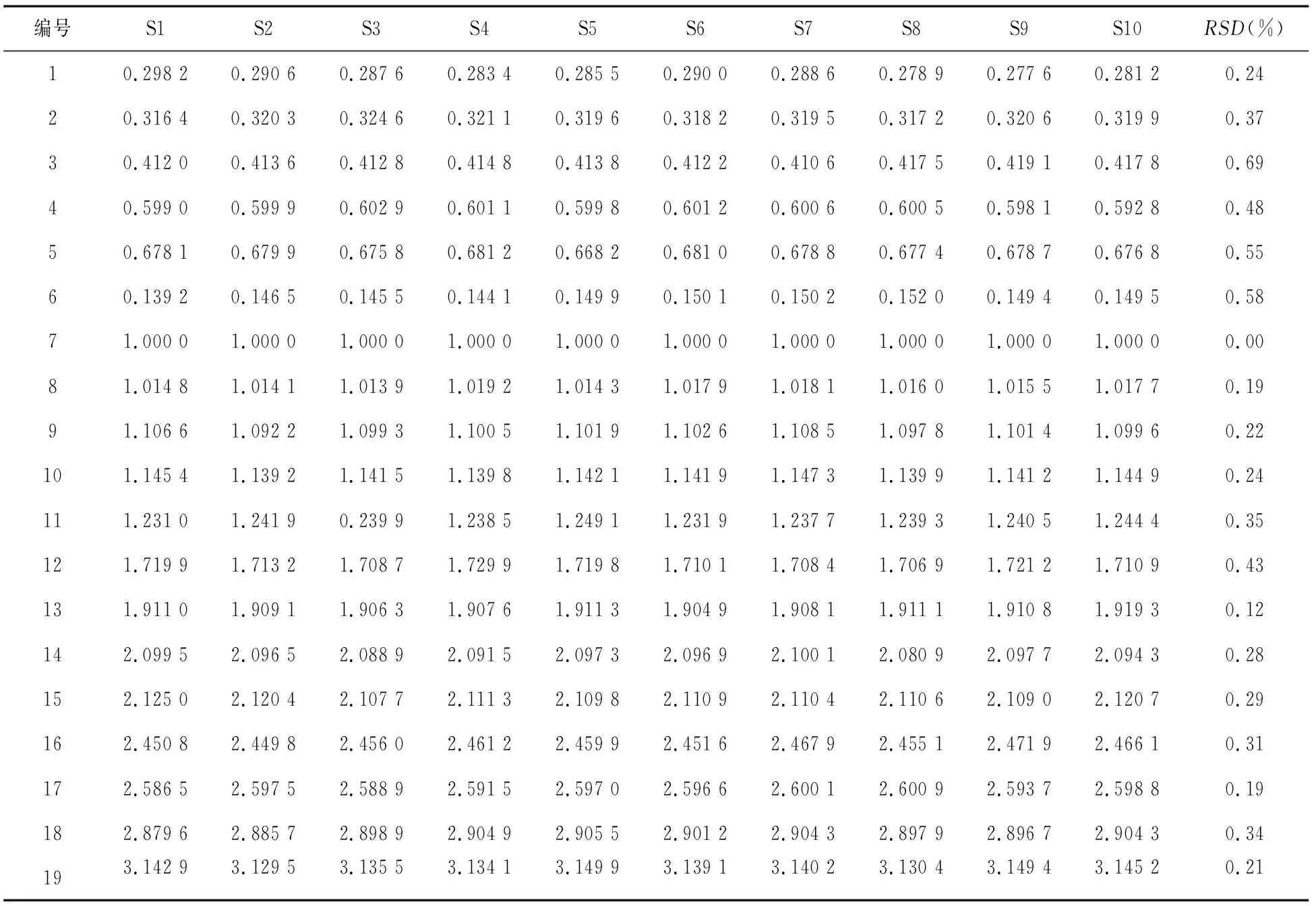
表3 虎杖解毒顆粒(無糖型)共有峰的相對保留時間
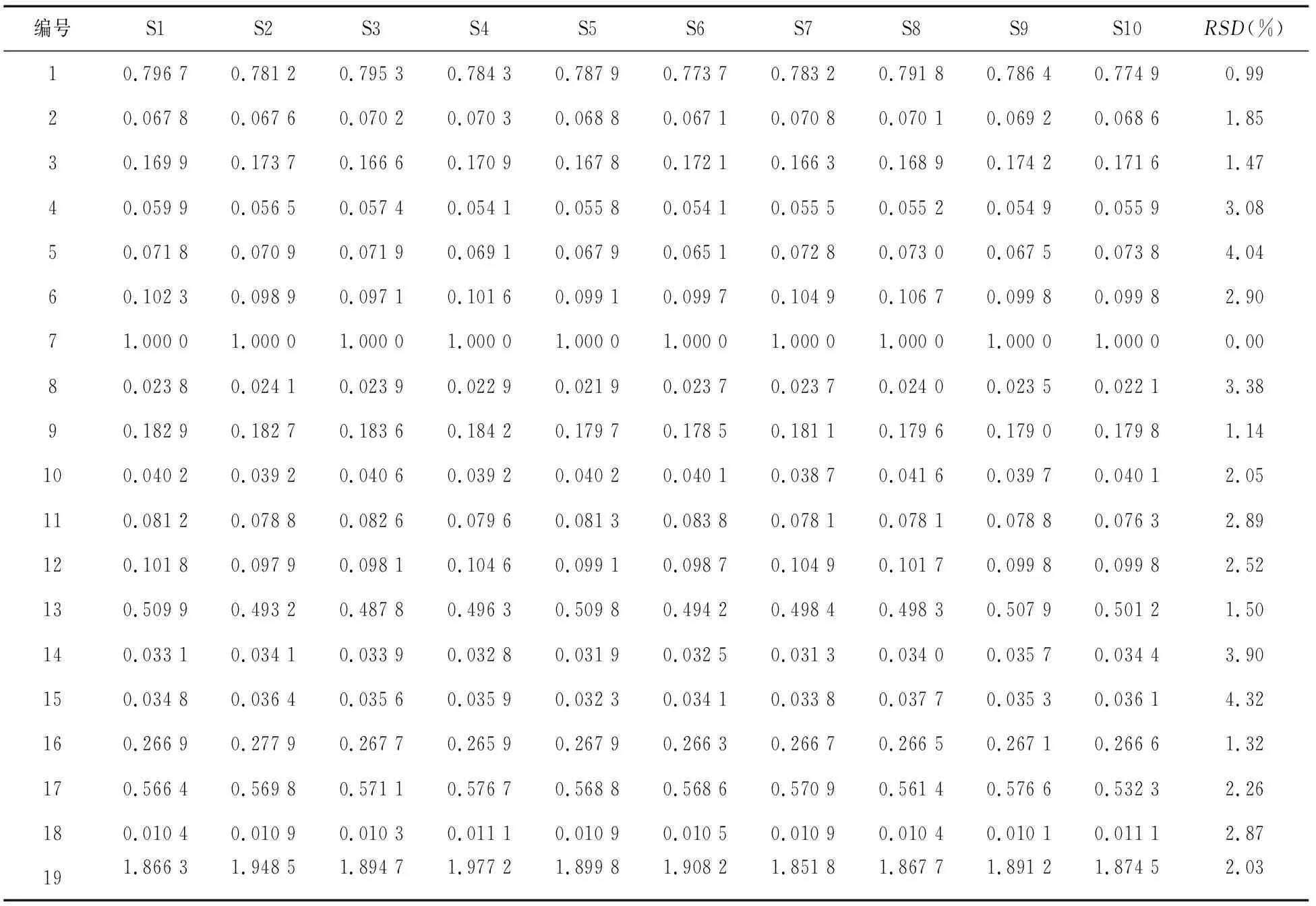
表4 虎杖解毒顆粒共有峰的相對峰面積

表5 虎杖解毒顆粒共有峰的相對保留時間
由表2~表5可知,在10個不同批次的虎杖解毒顆粒(無糖型)和虎杖解毒顆粒的指紋圖譜中,共有峰的相對保留時間RSD均小于1%,相對峰面積RSD均小于5%,表明2種制劑的工藝均穩定可行,指紋圖譜適應性良好。
2.5.2 指紋圖譜的相似度計算 將各10批供試品的色譜圖數據以AIA.格式導入中藥色譜指紋圖譜相似度評價系統(2012版),進行相似度評價[6]。結果10批虎杖解毒顆粒(無糖型)和虎杖解毒顆粒指紋圖譜的相似度平均值分別為 0.998和0.997,RSD值分別為0.32%和0.46%,符合指紋圖譜測定要求,表明2種供試品制劑的各批次間差異較小,質量穩定性較好。
3 結果與討論
3.1 色譜條件的優化 由于復方中藥制劑成分復雜,在一個波長下難以體現所有成分的特征,本研究在供試品溶液全波長掃描基礎上,重點考察250、300、360 nm波長切換掃描,結果顯示,在300 nm條件下,檢測到的色譜峰信息更全面,基線更平穩,故作為檢測波長[7-9]。
參考相關文獻對虎杖等藥材中相關成分的色譜條件研究[10-11],考察了不同提取溶劑(乙醇、水、甲醇)、不同柱溫(30、35、40 ℃)、不同超聲時間(30、60、90 min)、不同流動相體系(乙腈-水、乙腈-0.2%磷酸溶液、乙腈-0.05%磷酸溶液)對色譜結果的影響。結果表明,用甲醇超聲處理60 min,柱溫30 ℃,流動相乙腈-0.05%磷酸溶液,各色譜峰分離情況及峰形較好。由于供試液成分在圖譜前段重疊多、分離情況差,故對流動相前20 min降速,之后保持1 ml/min梯度洗脫。供試液中成分均在120 min前出峰,之后沒有相關色譜峰出現,故進樣時間定120 min。
3.2 虎杖解毒顆粒(無糖型)和虎杖解毒顆粒的圖譜分析和對照 虎杖解毒顆粒(無糖型)和虎杖解毒顆粒各批次的指紋圖譜相似度均大于0.98,說明創建的指紋圖譜的技術指標穩定可靠、重復性較好,能夠較好地全面反映制劑中化學成分的特征,并監控其成分及含量的變化。
對虎杖解毒顆粒(無糖型)和虎杖解毒顆粒指紋圖譜共有模式進行對照,其19個共有色譜峰的相對保留時間基本一致。結果表明,兩制劑的主要成分基本無變化,本研究為虎杖解毒顆粒(無糖型)研發的科學性、合理性提供了依據。
