VHL基因新突變致VHL綜合征伴腦膜瘤一家系分析
2021-12-14 09:33:32王楚楚齊光照張麗俠王志芳許莉軍鄭麗麗秦貴軍栗夏蓮
鄭州大學學報(醫學版) 2021年6期
關鍵詞:功能
李 沖,王楚楚,齊光照,張麗俠,王志芳,許莉軍,鄭麗麗,秦貴軍,栗夏蓮
1)鄭州大學第一附屬醫院內分泌及代謝科 鄭州450052 2)鄭州大學生命科學學院 鄭州450001 3)鄭州大學第一附屬醫院藥學部 鄭州450052
von Hippel-Lindau(VHL)綜合征是一種罕見的涉及多系統的常染色體顯性遺傳性腫瘤綜合征(OMIM 193300),發病率為1/50 000~1/36 000。VHL基因(ID:7428,https://www.ncbi.nlm.nih.gov/gene/7428)的體系或胚系突變導致多個器官發展成為良性、惡性腫瘤和囊腫,包括中樞神經系統血管網狀細胞瘤、視網膜血管網狀細胞瘤(HGB)、腎癌(RCC)或腎囊腫、嗜鉻細胞瘤(PHEO)、胰腺腫瘤或囊腫和生殖系統囊腫等病變[1-3]。VHL綜合征在65歲人群中的外顯率超過90%[4]。該病最常見的死亡原因是中樞神經系統的血管網狀細胞瘤和轉移性腎癌及其并發癥。突變個體發生神經系統病變的風險顯著增加,常見幕下(小腦、脊髓和腦干)神經母細胞瘤[5],出現在幕上的病變較為罕見。本研究對1例VHL綜合征合并右額葉腦膜瘤患者進行VHL基因變異家系分析,通過錯義突變蛋白功能預測軟件Polyphen-2和3D圖對該突變進行功能預測,結合文獻復習,探討腦膜瘤和VHL綜合征的關系。
1 對象與方法
1.1 病例資料患者,男,30歲,以“發作性心悸、頭痛、視物模糊19 a余,視物模糊再發3個月”為主訴于2020年3月收住鄭州大學第一附屬醫院內分泌及代謝科。19 a前(2001年)無誘因出現發作性心悸、頭痛、惡心,伴視物模糊,至當地醫院測血壓200/100 mmHg(1 mmHg=0.133 kPa)。眼底出血,腎上腺皮髓質功能未查,彩超示右腎上腺區實性占位(5 cm×2.6 cm×3.5 cm),行“右側腎上腺嗜鉻細胞瘤摘除術”。之后監測血壓均正常。15 a前(2005年)在當地醫院查腹部彩超和CT顯示左側腎上腺區實性占位,再次行左側腎上腺嗜鉻細胞瘤摘除術”。4 a前(2016年)在當地醫院查腹部彩超、增強CT和SPECT顯示右側腎上腺區、腔靜脈前門脈后嗜鉻細胞瘤可能,行“腹腔鏡下異位嗜鉻細胞瘤切除術”,病理診斷為(副主動脈旁)副節瘤。3個月前(2019年12月)再次出現左眼視物模糊,當地診斷為左眼底出血,腹部CT顯示腹膜后嗜鉻細胞瘤術后改變,左側腎上腺區多發病灶,考慮嗜鉻細胞瘤可能。自發病以來,神志清,精神可,大小便正常,體重無明顯變化。既往史、個人史、婚育史無特殊。體格檢查:除手術瘢痕外,未見異常。
1.2 家系概況及VHL基因檢測收集患者(先證者)外周血及病理組織切片。患者自述非近親結婚。父親及奶奶血壓高,服藥效果好。患者有1兄1弟1姐及2女,均體健,測血壓均正常。獲得患者及其家屬同意后收集5人的外周血樣本,進行VHL基因檢測。
1.2.1基因組DNA的提取 采用康為世紀生物科技有限公司的血液基因組DNA Mini試劑盒提取外周血DNA。采用南京諾唯贊生物科技有限公司的FastPure?FFPE DNA Isolation試劑盒提取病理組織切片基因組DNA。提取過程中的配套器材及操作流程嚴格按照說明書進行。
1.2.2VHL基因編碼區的擴增 用Primer 5.0軟件設計3對引物,采用南京諾唯贊生物科技有限公司提供的2×Taq Plus Master Mix Ⅱ(Dye Plus)試劑盒進行PCR擴增;引物序列見表1。PCR反應體系:2×Taq Plus Master Mix Ⅱ(Dye Plus)12.5 μL,上下游引物(10 μmol/L)各1 μL,基因組DNA模板(15~25 mg/L)1 μL,ddH2O補至25 μL。

表1 PCR引物序列
1.2.3PCR產物測序 PCR產物經瓊脂糖凝膠電泳后進行目的條帶純化,然后在鄭州普利萊醫學檢驗所實驗室進行測序。應用Sanger測序技術對患者外周血及病理組織切片進行VHL基因全部編碼區測序。使用Chromas及Sequencing Analysis Version 5.0對測序結果進行分析,測序結果與GenBank數據庫中序列比對。
1.2.4蛋白功能預測 利用軟件Polyphen-2(www.genetics.bwh.harvard.edu/pph2)對錯義突變蛋白進行功能預測。
2 結果
2.1 實驗室、影像學和病理學檢查結果患者的血尿糞常規、肝腎功、電解質、甲狀腺功能、促甲狀腺激素(TSH)、腎上腺髓質功能檢查均未見異常。全天血壓動態變化呈弱杓型曲線。眼底檢查見圖1A,全腹部CT平掃+增強檢查結果見圖1B,腎上腺髓質SPECT/CT融合顯像結果見圖1C,頭顱MRI平掃與增強結果見圖1D。患者于2020年3月在我院神經外科行“右額葉腫塊切除術”。術后病理顯示(右額葉占位)腦膜瘤,WHOⅠ級。免疫組化結果:CK(-),S-100(-),EMA(-),SSTR2(+),PR(部分+),GFAP(-),Ki-67(2%+),CD34(血管+),ERG(血管+),NSE(-),Inhibi-a(-),STAT6(-),CgA(-),Syn(-),D2-40(血管+)。

A:眼底照相顯示左眼視盤表面可見橘紅色圓形隆起病灶,左眼眼底熒光血管造影提示視網膜毛細血管瘤;B:腎上腺CT顯示左側腎上腺區多發病灶;C:腎上腺髓質SPECT/CT融合顯像,靜脈注射顯像劑131I-MIBG后,左側腎上腺可見軟組織結節影,部分放射性分布濃聚,大者約1.3 cm×1.5 cm,CT值約41 Hu;腹膜后可見軟組織結節影放射性分布濃聚,大小約1.4 cm×1.9 cm,CT值約27 Hu;D:頭顱MRI顯示右側額葉見類圓形長T1長T2信號,內見多發小圓形長T1長T2信號,T2 Flair序列部分呈高信號,高B值DWI可見點片狀擴散受限高信號,病變大小約26 mm×26 mm×21 mm(前后徑×左右徑×上下徑)圖1 患者影像學資料
2.2 VHL基因檢測結果及分析患者血液和病理組織標本的測序結果均提示患者攜帶1個VHL基因的雜合錯義變異(c.284C>G),該變異導致第95位編碼氨基酸殘基由脯氨酸Pro變為精氨酸Arg(p.Pro 95 Arg)。患者父母及一個女兒均未攜帶該變異,提示該變異為新生變異。另一個女兒為該變異雜合子攜帶者,其測序結果見圖2A。
c.284C>G變異未被ClinVar數據庫收錄,但收錄在HGMD數據庫中,并在1例嗜鉻細胞瘤患者中有過報道[6]。該變異附近氨基酸殘基的改變(p.Asn 90 Ile;p.Gly 93 Cys;p.Gly 93 Arg;p.Gly 93 Asp;p.Tyr 98 His;p.Tyr 98 Cys)與VHL綜合征的致病或可能致病均已有報道[7-9],表明該變異位于熱點突變區。此外,該變異頻率在ExAC、GenomAD、1000G參考數據庫中未見收錄,不屬于多態性變化。c.284C>G編碼蛋白p.Pro 95 Arg所在區域片段的物種保守性分析顯示p.Pro 95在智人、恒河獼猴、老鼠、狗、非洲大象和雞這些物種中高度保守(圖2B),位于VHL第一外顯子(圖3A)。經過錯義突變蛋白功能預測軟件Polyphen-2預測,該突變對VHL蛋白造成顯著影響,預測的氨基酸突變為probably damaging,評分為0.994(圖3B)。在蛋白質3D圖上,突變位點c.284C>G位于VHL的功能區,與缺氧誘導因子(HIF)結合,具有非常重要的功能(圖3C)。結合在家系驗證中其父母未攜帶該變異的情況,根據ACMG指南判定該變異為可能致病,會對基因或基因產物造成有害的影響。

A:紅色箭頭示變異位置,先證者(Ⅱ:1)病理樣本及血液樣本測序結果均提示攜帶變異(c.284C>G);父(Ⅰ:1)、母(Ⅰ:2)、女兒1(Ⅲ:1)未攜帶該變異;女兒2(Ⅲ:2)攜帶該變異;B:VHL:p.Pro 95 Arg所在編碼蛋白質區域片段的物種保守性分析顯示p.Pro 95在物種間高度保守圖2 先證者VHL基因測序圖
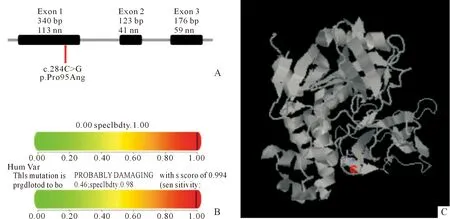
A:c.284C>G位于VHL第一外顯子;B:Polyphen-2預測顯示該突變對VHL蛋白造成顯著影響,預測的氨基酸突變為probably damaging,評分為0.994;C:在3D圖上,其蛋白產物位于VHL的功能區,與HIF結合,具有非常重要的功能圖3 VHL基因 c.284C>G突變的蛋白功能預測
3 討論
VHL是一種重要的腫瘤抑制因子,參與HIF的泛素化和降解,在氧調控基因表達中起著重要作用。VHL位于染色體3p25.3,包括3個外顯子,編碼蛋白是蛋白質復合物的組成部分,包括延伸蛋白B、延伸蛋白C、Cullin-2和具有泛素連接酶活性的E3。VHL突變導致蛋白失活,從而使下游底物上調,促進一系列致癌基因的表達,是多器官腫瘤發生的重要機制[10]。
VHL綜合征具有典型的基因型-表型相關性。根據是否合并嗜鉻細胞瘤,分為兩型:1型無嗜鉻細胞瘤,常由VHL外顯子缺失、截斷、移碼或無義突變引起;結合是否合并腎癌,分為1A型(合并腎癌)和1B型(無腎癌)。2型包含嗜鉻細胞瘤,多由VHL錯義突變引起;進一步又可分為腎癌風險低的2A型、腎癌風險高的2B型和只有嗜鉻細胞瘤表現的2C型[1,11]。
本文報道的VHL綜合征患者11歲因高血壓陸續發現左側腎上腺和腹膜后嗜鉻細胞瘤,此次因左眼視物模糊入院,完善FFA(眼底熒光血管造影)后發現左眼視網膜毛細血管瘤,綜合考慮可能為VHL綜合征,進一步對血液和嗜鉻細胞瘤病理組織進行基因檢測,顯示該患者攜帶VHL雜合錯義突變c.284C>G,導致第95位氨基酸由脯氨酸Pro變為精氨酸Arg(p.Pro 95 Arg),為胚系突變。經不同物種氨基酸序列比對,第95位脯氨酸在多個物種中具有高度保守性。VHL功能缺失等同于細胞缺氧狀態,這可能是導致該患者發生多種腫瘤的分子機制。
進一步檢查發現右側大腦額葉占位,術后病理顯示腦膜瘤。經過文獻檢索,腦膜瘤與NF2雜合缺失有關,在VHL綜合征中非常罕見[12],近年來僅有少數VHL綜合征合并腦膜瘤的病例報道,Kanno等[13]報道的一例VHL綜合征2B型合并腦膜瘤患者在VHL第三外顯子685位胞嘧啶突變為鳥嘌呤(685C>G)導致158位亮氨酸突變為纈氨酸(L158V)(胚系突變)。Santarpia等[14]描述一例26歲VHL綜合征2C型患者,先后經歷了雙側腎上腺嗜鉻細胞瘤、腹主動脈和膀胱副神經節瘤切除術,后來發現右側額葉腦膜瘤,基因檢測顯示VHL695G>A導致161位精氨酸突變為谷氨酰胺(R161Q)。在本研究中,該患者為VHL綜合征2C型,存在VHL第一外顯子的雜合錯義突變(c.284C>G),通過Polyphen-2和蛋白結構3D圖對錯義突變c.284C>G的功能進行預測,提示該突變是功能突變,能顯著影響VHL的活性,與臨床表型關系密切,但是否導致其罹患右側額葉腦膜瘤尚不清楚。
隨后我們對患者的家系成員進行突變位點的檢測,患者一8歲女兒攜帶VHL c.284C>G,目前并未發現有相關腫瘤及囊腫的發生。根據國內外指南,對于無癥狀VHL基因突變攜帶者目前尚無有效的干預腫瘤發生的手段,只能進行密切監測[15]。由于視網膜腫瘤的發生風險是在青少年時期,我們正在對該女進行定期隨訪、預防性監測,待成年后進行遺傳咨詢,以期早期發現腫瘤,及早治療,提高其生活質量和預期壽命。
VHL突變的多樣性,相同突變位點外顯率和發病年齡的顯著差異給VHL綜合征的診治帶來了巨大的挑戰[16]。Iida等[17]對存在VHL基因695G>A(R161Q)突變的VHL綜合征2A型先證者的16位家庭成員進行檢測,發現8例攜帶相同突變位點的基因,其中一例表現為胰腺神經內分泌腫瘤,無嗜鉻細胞瘤,同卵雙胞胎中的一例表現為巨大的嗜鉻細胞瘤和視網膜母細胞瘤,而另一例則無任何表型。目前尚不能通過基因檢測來預測患者可能出現的表型,比如哪個器官會受累,發生風險的高低,腫瘤在特定生命階段(幼兒期、妊娠期等)的特殊發生,或腫瘤生長的速度等。VHL基因的胚系突變與產生的臨床表型之間的相關性取決于突變的VHL下調HIF途徑活性的能力,此外,年齡、性別、基因型、妊娠、腫瘤的位置等都可能影響患者的臨床表型[18]。近年來,許多與HIF無關的VHL功能被發現,這進一步提示了VHL綜合征表型調控機制的復雜性。
總之,我們發現了VHL綜合征中一個新的胚系突變VHL c.284C>G,對VHL的功能可能造成顯著影響,該突變與腦膜瘤發生發展的相關性是我們未來進一步研究的焦點。同時,本課題組正在開展對無癥狀VHL突變攜帶人群進行密切隨訪和實施潛在干預的臨床研究,以提高VHL綜合征患者的生活質量,改善其預后。
猜你喜歡
鐘表(2023年5期)2023-10-27 04:20:44
中華詩詞(2022年6期)2022-12-31 06:41:24
當代陜西(2021年21期)2022-01-19 02:00:26
中學生數理化(高中版.高考數學)(2020年1期)2020-02-20 13:23:44
經濟技術協作信息(2018年11期)2019-01-14 03:07:20
中國科技論壇(2017年7期)2017-07-25 08:49:53
制造技術與機床(2017年3期)2017-06-23 08:11:33
媽媽寶寶(2017年2期)2017-02-21 01:21:24
國際漢語學報(2016年1期)2017-01-20 08:21:20
中國中醫藥現代遠程教育(2014年22期)2014-03-01 04:32:55
