1例早發腦型肝豆狀核變性家系ATP7B基因突變和臨床表型分析
2021-12-23 10:52:46錢南南王久香楊文明
中風與神經疾病雜志 2021年11期
錢南南, 王久香, 裴 培, 楊 悅, 楊文明
肝豆狀核變性(Wilson’s Disease,WD,MIM 277900)是一種常染色體隱性遺傳的以銅代謝紊亂為特征的代謝性疾病,主要表現為肝功能異常、神經或精神損害、角膜可見K-F環(Kayser-Fleischer ring)甚至腎損傷。臨床上,該病主要分為肝型(肝功能損傷)、腦型(神經精神異常)和混合型(二者都異常)。患者一般在5~35歲發病,兒童以肝型為主,腦型患者少見[1,2]。目前發現WD是由ATP7B基因突變所導致,我們對1例中國漢族早發型腦型WD家系進行ATP7B變異鑒定,以明確其遺傳學病因,為該家系的疾病診斷提供遺傳學證據,對家系其他成員的再發風險進行指導。
1 對象與方法
1.1 對象 先證者,女,13歲。2017年7月份漸出現口角流涎、言語不清,寫字慢、字體歪斜。就診于當地衛生所考慮“胃炎”,予口服藥治療未見好轉(具體用藥不詳)。發病16 m開始出現左手不自主抖動,記憶力明顯減退。患者雙眼角膜K-F環陽性,腦部MRI顯示雙側基底節區、黒質對稱性異常低信號(見圖1),24 h尿銅 621.7 μg/d,ALT、AST、GGT和AKP正常,CER 0.093 g/L(參考值0.2~0.6 g/L),考慮“肝豆狀核變性”,予排銅、保腦等綜合治療后口角流涎及左手不自主抖動較前好轉。患者4歲時患過病毒性腦炎。

表1 ATP7B外顯子及其側翼區PCR擴增引物序列
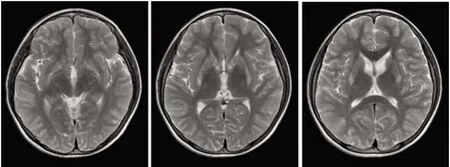
先證者腦部MRI顯示雙側基底節區、黒質對稱性異常低信號圖1 先證者頭部MRI
本研究遵循《赫爾辛基宣言》,經中國合肥安徽中醫藥大學第一附屬醫院倫理委員會批準,本研究參與者均簽署知情同意書。
1.2 方法
1.2.1 基因組DNA提取 EDTA抗凝管采集先證者及家系其他成員的外周血3 ml(見圖2A),全血基因組DNA提取試劑盒(北京天根生化科技有限公司)提取基因組DNA,并用分光光度計(Eppendorf,德國)檢測其濃度和質量。
1.2.2 基因測序 利用Primer Premier 5 軟件設計擴增包含ATP7B基因外顯子及其外顯子內含子連接區引物,由武漢擎科生物有限公司合成(見表1)。PCR體系(50 μl):5X PrimeSTAR緩沖液(含Mg2+)10 μl (Takara)、基因組DNA 100 ng、上下游引物各0.2 μmol/L、dNTP Mixture 0.2 mmol/L(Takara),PrimeSTAR DNA polymerase 0.5U(Takara),用H2O補足至50 μl體系。PCR反應條件:98 ℃預變性5 min;98 ℃ 變性10 s;55 ℃~60 ℃退火5 s,72 ℃延伸1 min,共35個循環,最后72 ℃延伸10 min,4 ℃保存。PCR產物送武漢擎科生物有限公司用ABI PRISM 3100測序儀測序。測序結果與NCBI數據庫中的ATP7B基因序列進行比對(見圖2B-2E)。
2 結 果
ATP7B基因測序結果:外顯子直接測序結果顯示,先證者ATP7B基因存在c.2333G>T和c.2975C>T復合雜合錯義變異(見圖2C),分別導致ATP7B第778位精氨酸變成亮氨酸,第992位脯氨酸變成亮氨酸。經檢索HGMD數據庫、dbSNP、千人基因組等數據庫證實,c.2333G>T和c.2975C>T均為已知WD致病位點。先先證者母親攜帶c.2333G>T(p.Arg778Leu)突變(見圖2B),先證者父親攜帶c.2975C>T(p.Pro992Leu)突變(見圖2C)。先證者的弟弟攜帶c.2333G>T(p.Arg778Leu)突變(見圖2E)。
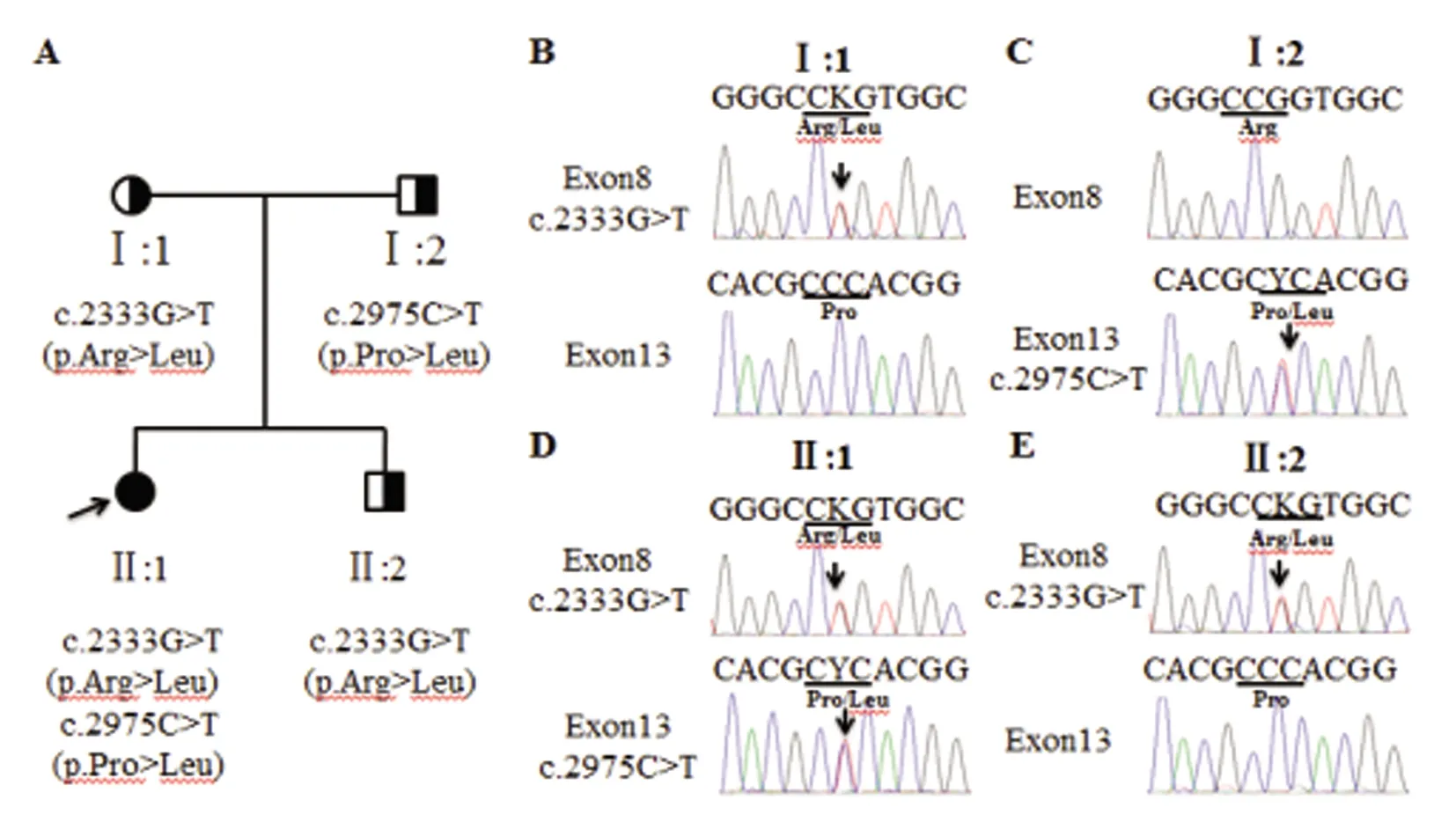
A:先證者家系圖,箭頭指示先證者;B~E:ATP7B測序結果(正向),箭頭指示變異位點圖2 先證者家系及測序結果圖
3 討 論
WD是由于體內銅離子異常累積導致的一種罕見隱形遺傳病。ATP7B是目前已知的WD致病基因,其定位于13q14.3,編碼含有1465氨基酸殘基的跨膜蛋白。銅離子的代謝障礙導致其在腦部、肝臟、眼底甚至腎臟累積,導致患者出現肝硬化、神經精神紊亂、角膜K-F環甚至腎損傷。因此早診斷、早治療可減少患者肝、腦等組織的損傷,延緩病情進展,明顯提高WD患者的生存質量,最終回歸家庭和社會。患者自出生即存在銅代謝障礙,一般3歲以后才開始出現癥狀,臨床表現各異且大部分為非特異性,因此早期診斷仍較為困難[3]。本文先證者發病初期口角流涎、言語不清,曾考慮胃炎,外院治療后未見好轉。發病4 m后,流涎、言語不清加重,開始出現左手不自主抖動。
目前ATP7B基因已經發現有600多個WD致病突變位點。H1069Q是目前中歐最常見的WD突變位點,在波蘭、羅馬尼亞,奧地利等中歐國家的等位基因頻率為30%~60%[4]。R778L是日本、韓國、中國等亞洲患者的熱點變異位點[5]。R778L等位基因在韓國WD患者的攜帶率約為 37.9%、日本約為27%,中國約28%~44%[4,6,7]。c.2975C>T(p.Pro992Leu)突變位于ATP7B第8外顯子,蛋白的跨膜結構域。2013年,有學者報道了1例9歲的中國WD男童,攜帶與本文先證者相同的突變位點ATP7B c.2333G>T和c.2975C>T[8]。該患者神經精神正常,本文中的先證者有明顯的神經精神癥狀;角膜K-F環陰性,本文患者陽性;肝功能指標ALT(137 IU/L)、AST(106 IU/L)明顯升高,本文先證者ALT(16 U/L,參考值5~35 U/L)、AST(22 U/L,參考值5~35 U/L)。有多位學者嘗試尋找患者基因型和發病年齡、性別、臨床表型的關系,但是沒有明確的結論[9~11]。最新研究顯示ATP7B DNA甲基化可能與患者臨床表型相關[12]。此外先證者4歲時曾患有病毒性腦炎,是否與患者的早期發病相關亦未可知。
先證者的母親攜帶ATP7B c.2333G>T突變,父親攜帶c.2975C>T突變。先證者從雙親遺傳到ATP7B c.2333G>T和c.2975C>T符合雜合突變,結合患者臨床癥狀、生化檢查及體征,診斷WD明確。通過對先證者的弟弟ATP7B 基因21個外顯子和外顯子內含子銜接區的測序發現其只攜帶c.2333G>T突變,提示其弟弟的發病可能性不大,可密切隨訪觀察。
我們報告了一例早發型腦型WD家系,明確該家系WD發病的遺傳學基礎是ATP7B c.2333G>T和c.2975C>T,并對先證者的弟弟進行突變檢測,分析其患病的可能性。
