致病氨基酸變異預測的新型融合模型
2022-01-26 12:42:52邵愛斌
電子科技大學學報 2022年1期
邵愛斌,楊 洋*
(1. 蘇州大學計算機科學與技術學院 江蘇 蘇州 215006;2. 軟件新技術與產業化協同創新中心 南京 210000)
隨著測序成本的大幅降低,測序方法在科研和臨床醫療中被廣泛使用,由此產生了大量測序信息,也包括了越來越多的變異信息。基因變異導致的氨基酸變異可通過多種方式影響蛋白質的結構和功能。當變異發生在蛋白質的某些關鍵部位,如催化部位或配體相互作用表面,可能導致蛋白質折疊、結構不穩定或蛋白質聚集,進而導致疾病。為實現個性化醫療,追溯疾病發生的機理,預測氨基酸變異的致病性具有很高的研究價值。
與實驗方法相比,計算方法具有預測成本低、效率高的突出優勢。近年來,研究者們提出并不斷改進了多種相關預測模型。其中,PolyPhen-2[1]面對不同預測任務靈敏性的需求,構建了HumDiv和HumVar 兩個數據庫,運用樸素貝葉斯預測變異致病性。CADD[2]使用支持向量機算法,整合了63 種基因注釋,從而得到C 分數來預測致病性。PON-P2[3]利用基因本體(gene ontology, GO)等特征訓練,采用自抽樣的方式計算置信度。DEOGEN2[4]用可視化的方式提供了每個預測的結果、相關蛋白質背景和起源。MetaSVM[5]開發了基于支持向量機的元分析框架,框架中SVM 的目標函數由鉸鏈損失和稀疏組套索組成。ClinPred[6]首次使用了ClinVar[7]數據庫,并訓練了兩個模型,分別基于隨機森林和XGBoost 來獲取最高預測結果。PrimateAI[8]結合6 個非人類靈長類動物物種和人類的變異,共收集到38 萬條變異數據,訓練了一個包含36 層卷積神經網絡的深度學習模型。
隨著當前可采集的變異數據量的增加,構建一個新的融合模型以提高預測性能變得可行。因此,本文嘗試使用深度學習方法從蛋白序列中提取出一些特征,將這些特征與提取并篩選的有效生物特征融合,作為模型的輸入,并構建融合模型訓練,旨在達到較高的預測性能。
1 數據來源
本文所使用的數據包括人類、動物和植物蛋白序列中的氨基酸變異樣本,如表1 所示。其中,人類致病變異取自VariBench[9-10]數據庫和ClinVar[7]數據庫,共有17631 個。按致病變異與無害變異大約1∶1 的比例,從ExAC[11]數據庫獲取共18494 個人類無害變異。對于動物變異數據:1)收集OMIA[12]數據庫中有“likely causal variants”標記的變異;2)從文獻[13]獲得其他哺乳動物(狗、鼠和牛)的變異。動物致病氨基酸變異共317 個,中性變異共312 個。進一步從文獻[14]取得植物變異數據集,其物種包含擬南芥、水稻和豌豆,數據集由3236 個有害變異和1899 個中性變異構成。
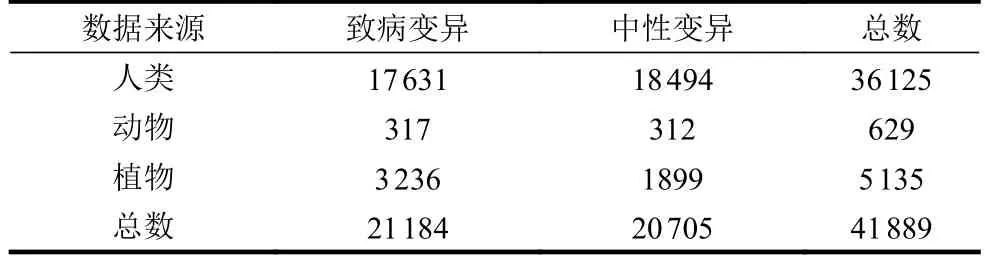
表1 實驗數據來源和分布 個
由于某些蛋白序列過長,影響預測性能,本文篩選出長度不超過1500 個的蛋白質序列,共9980條,包含了35179 個氨基酸變異,其中致病變異18521 個,無害變異16658 個。
2 方 法
本文所構建的新型融合模型工作流程如圖1所示。
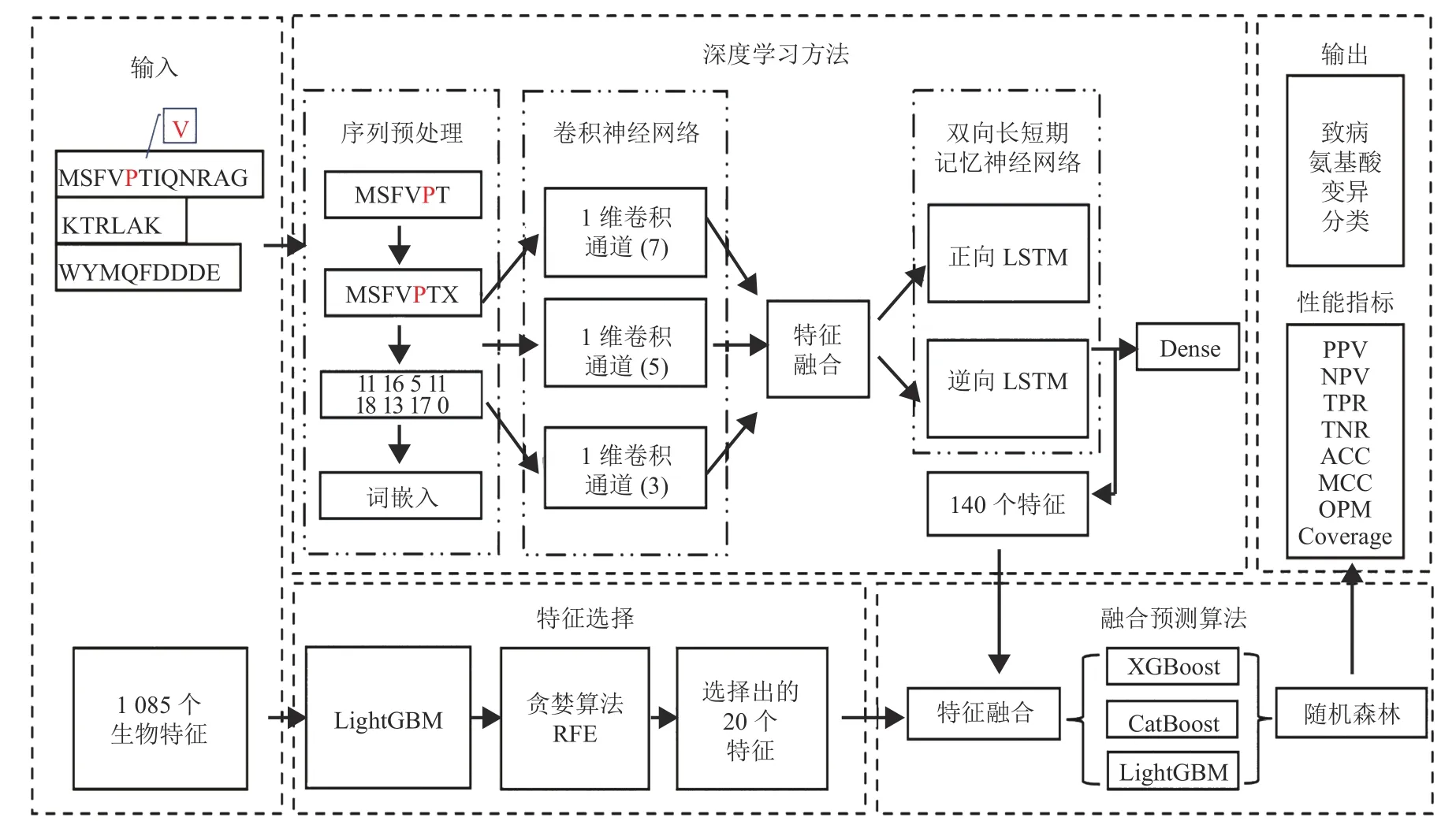
圖1 致病氨基酸變異預測的新型融合模型的工作流程
2.1 生物特征提取
綜合文獻[3, 15]中的特征,本文共提取了1085個可能與氨基酸變異致病性有關的生物特征,包括617 個氨基酸特征、436 個變異類型特征、25 個鄰域特征、2 個進化保守特征、3 個蛋白質特征以及GO 頻率對數比特征(logical rate, LR)和功能位點特征(functional site, FS)。
617 個氨基酸特征通過AAindex 表[16]提取,對于每個變異,計算氨基酸變異前后得分的差異。
436 個變異類型特征通過兩個矩陣獲得。其中,400 個特征來自20×20 的矩陣,矩陣的兩個維度分別代表變異前和變異后的氨基酸。另外36 個特征來自代表氨基酸物理和化學性質的6×6 矩陣[17]。
25 個鄰域特征表示變異位點的序列位置特征。20 維向量在23 個位置,即變異位點前后11個位置的窗口中統計每種氨基酸類型的頻數[18]。此外,在23 個位置的鄰域窗口中統計了5 種氨基酸類別,包括非極性、極性、帶電、帶正電和帶負電的頻數。
使用DIAMOND[19]將每個蛋白質序列在SwissProt[20]中進行同源序列比對,找到同源序列并統計命中數。比對后,使用SIFT 4G[21]計算每個變體的SIFT 分數。
3 個蛋白質特征包括蛋白質序列的長度,變異是否發生在序列的第一位氨基酸,以及變異在序列中的位置。
利用樸素貝葉斯算法計算特征LR 和FS 如下:
1)使用R Bioconductor 工具GO.db(https://bioconductor.org/packages/GO.db/),從AmiGO[22]與QuickGO[23]中收集蛋白質的所有GO 術語以及這些術語對應的全部上層GO 術語,并過濾以避免重復。然后,在數據集中對收集到的GO 術語分別統計兩個類別,即致病和無害的頻數,計算該蛋白GO 的LR 如下:
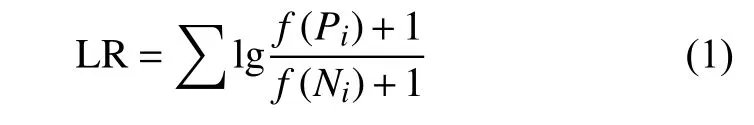
式中,f(Pi)是第i個GO 術語在致病集的頻數;f(Ni)是第i個GO 術語在無害集中的頻數。為了避免分數無意義,頻數都加1。如果一個蛋白質沒有GO 術語,則LR 為0。
2)根據UniProtKB/SwissProt[20]查找變異位點的Site 術語,以類似LR 的計算方法得到該蛋白Site 的功能位點特征。
2.2 生物特征選擇
特征選擇也稱屬性選擇,通過從當前全部特征中篩選出部分特征的方式,達到模型某項指標最優化的目的。較多的特征中可能存在無關特征和冗余特征,這也許會導致維度災難,使得模型的性能下降,訓練的時間開銷增加。當特征較多而樣本數量較少時,模型往往容易出現過擬合的問題。為了避免特征冗余和模型過擬合的風險,減少算力資源浪費,增加模型可解釋性,需要篩選出與分類性能密切相關的生物特征。
參照文獻[1, 3]的特征選擇方法,本文使用LightGBM 算法結合遞歸特征消除(recursive feature elimination, RFE)方法[24]進行特征選擇。特征選擇的過程是以全部特征構建模型,為每個特征分配權重,并去除權重最小的特征,然后在剩余的特征上重復上述操作,直到達到預設的特征數量。
2.3 通過神經網絡提取特征
隨著數據量增加,有望通過深度學習方法提取出一些沒有明確生物意義但對分類有幫助的特征。本文構建的深度學習網絡主要包括兩個部分:卷積神經網絡(convolutional neural networks, CNN)和雙向長短期記憶神經網絡(bi-directional long-short term memory, Bi-LSTM)。
CNN 提取特征的基本思路是通過不斷平移卷積核,對原始矩陣的每個元素進行卷積操作,從而提取到某些細節特征。卷積核的感受野與卷積核大小有關,感受野不同,卷積網絡能提取到的特征細節也不一樣。本模型設置了多條卷積線路,不同線路對應不同卷積核。通過融合多種卷積核的卷積結果,可以提取到較全面的特征。本模塊使用了多層的小卷積核,代替少層的大卷積核。在相同大小感受野的前提下,能夠縮小計算量,提高訓練效率。對于每層卷積提取的特征,再利用激活函數進行非線性變換,以提取更具有泛化性的特征。
特征提取中,本文進一步使用了Bi-LSTM,其構造中有記憶門和遺忘門,經過訓練,能夠選擇記憶和遺忘某些信息,探索到CNN 輸出的特征向量中距離較遠的關系。Bi-LSTM 由正向LSTM 和逆向LSTM 組成,氨基酸變異位點特征同時與該位點前后序列都有關。經過特征累計,Bi-LSTM能夠提取出位點前后向的雙向特征。
2.4 融合預測算法
提取并篩選出的生物特征與深度學習提取到的特征以拼接的形式相融合,作為融合預測算法的輸入。融合特征與預測結果之間的關系比較復雜,多個模型的融合或許能取得較好的性能。不同的模型從不同的角度去觀測數據。堆疊法(stacking)可以整合多個模型觀測數據的角度,相互取長補短,能夠得到一個更加全面和優秀的結果。LightGBM[25]采用了基于梯度的單邊采樣,留下梯度大的樣本,隨機抽樣梯度小的樣本,來分割數據樣本。XGBoost[26]通過直方圖算法,將全部樣本按照某個特征分成離散區域,并根據這些區域確定最佳分割。CatBoost 采用貪婪策略,整合不同類別型特征得到新的特征,能夠直接處理類別型特征。所以,XGBoost、CatBoost 和LightGBM 這3 種提升boosting 算法各有特點。
本融合模型選擇這3 個算法作為第一層模型,訓練并保留預測結果,得到3 列新的特征表達。第二層隨機森林用得到的特征表達進行訓練,得到最終的分類結果。以此達到平衡各算法的優缺點,提高分類性能的目的。
2.5 模型評價
本文引入了一些評價指標對模型進行評估,來直觀辨別分類性能的優劣,其中包括陽性預測值(PPV)、陰性預測值(NPV)、敏感性(TPR)、特異性(TNR)、準確性(ACC)、馬修斯相關系數(MCC)和總體績效指標(overall performance measure, OPM)。這些評價指標的數學定義如下:
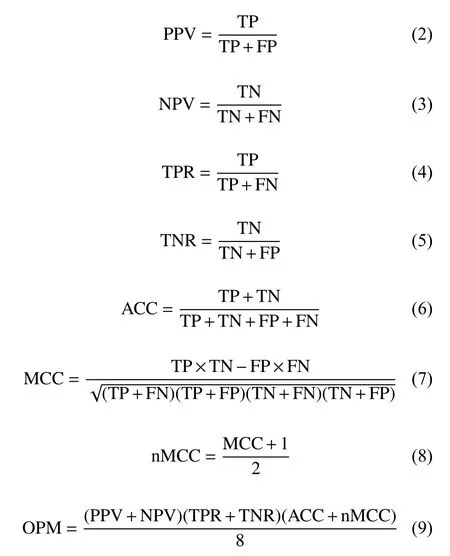
MCC 在TP、TN、FP 和FN 基礎上評價二元分類性能,其值在?1~+1 之間,值越大分類性能越好。OPM 由PON-P2[3]定義,將MCC 歸一化為0~1 值(nMCC),結合PPV、NPV、TPR、TNR和ACC,如式(9),兩兩之和為棱長作長方體,長方體體積的1/8 作為OPM 的值。OPM 的取值范圍為0~1,值越接近1,分類性能越好。
3 實驗結果
在數據集中隨機選取90%的變異樣本作為訓練集進行10 重交叉驗證。實驗用的部分數據和代碼已經上傳GitHub (https://github.com/s1mpleCN/PONDML)。
3.1 生物特征篩選結果
參照文獻[1, 3]的特征篩選經驗,本文用RFE 算法分別篩選了100、50、20、10 個特征進行比較。其交叉驗證結果如表2 所示,其中僅基于20 個生物特征,預測準確性ACC 即可達到89.7%,MCC 為79.4%,OPM 為72.2%。綜合考慮特征數量和模型性能的平衡,本文最終選擇了篩選出的20 個生物特征來進行特征融合。
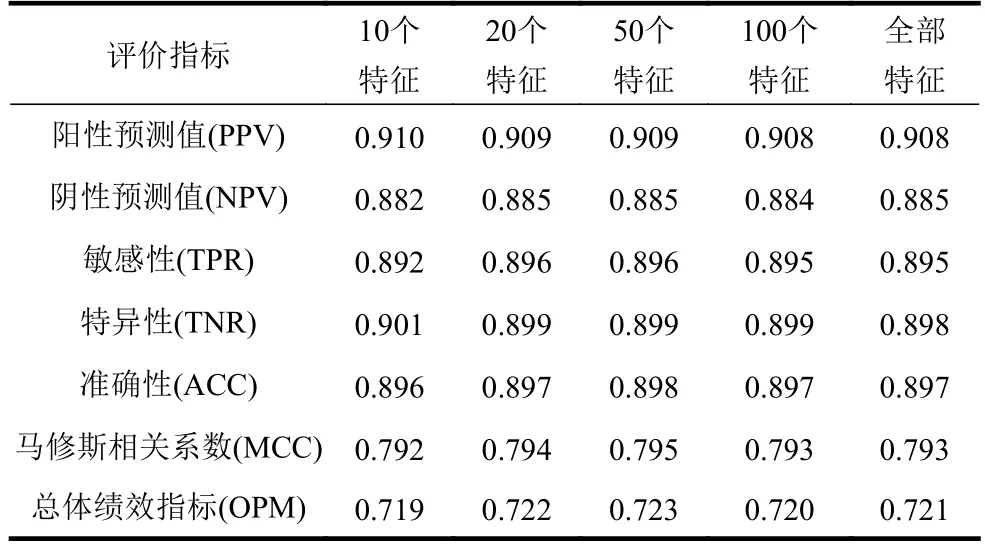
表2 篩選不同數量特征的CV 結果
3.2 通過神經網絡提取特征的結果
考慮到原始蛋白序列是由英文字母組成的,無法直接用深度學習方法訓練。借鑒自然語言處理的做法,將原始蛋白質序列按照氨基酸單字母縮寫的順序從小到大進行編碼[27]。神經網絡中的Bi-LSTM要求輸入的序列長度限制為固定長度,在長度短的序列后面填充0,使序列最終長度均為1500 個氨基酸。通過詞嵌入方法將序列中每個數字編碼,轉化為一個實數向量。本文設置嵌入參數為128,輸出為1500×128 的矩陣。
本文構建的CNN 使用了一維卷積函數,只需要定義核大小的一個參數,另一參數默認取詞嵌入部分設置的長度128。CNN 共構造了3 條卷積路線,分別對應3 種大小的卷積核(7×128、5×128和3×128)。每條路線中都包含5 個卷積層,每個卷積層之后都進行最大池化操作,從而提取出更顯著的特征。將3 條路線提取的特征以拼接的方式融合,作為Bi-LSTM 的輸入。經過實驗比較,Bi-LSTM 層的輸出參數設為70 時,預測性能最好。此時,正向LSTM 和逆向LSTM 都可以提取到70 個特征。
在構建的神經網絡中加入丟棄層(dropout),每次隨機選擇訓練過程中的部分神經元進行計算,避免模型過擬合。網絡中加入批標準化層(batch normalization),可以避免梯度爆炸,加快迭代速度。綜合考慮模型的準確性和泛化性,利用早停機制(early stopping),共進行30 次迭代。深度學習網絡參數詳見表3。

表3 深度學習網絡中的參數
神經網絡訓練的模型性能如表4 所示,其準確性ACC 為84.9%,MCC 為69.7%,OPM 達到61.2%。
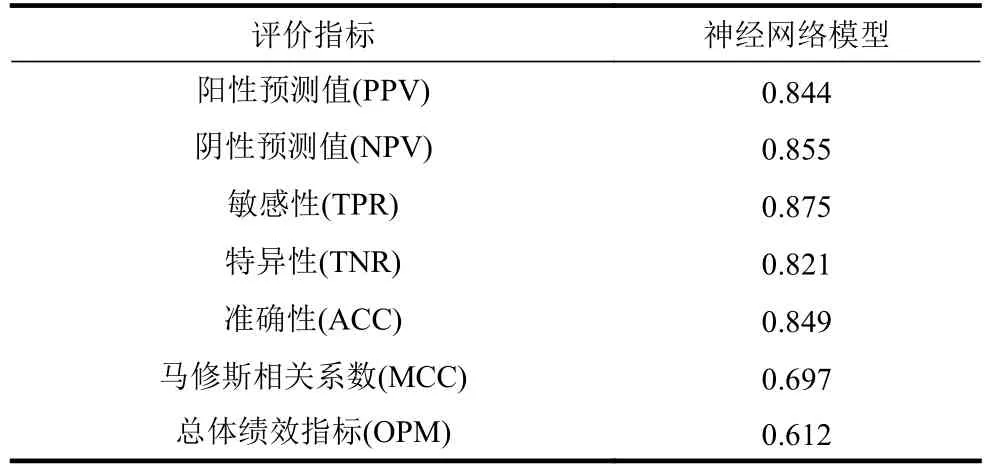
表4 神經網絡訓練的模型性能結果
3.3 融合模型交叉驗證性能結果
如圖2 所示,本融合模型將每次交叉驗證的訓練集隨機分成3 份,分兩層進行訓練。首先選擇XGBoost,每一次用其中兩份數據預測剩下的1 份數據以及驗證集的數據,保留得到的預測結果。每份數據都預測一次,共可以保留3 份訓練集的預測結果和3 份驗證集的預測結果。整合訓練集的預測結果作為第二層的一列新的特征表達。將3 份驗證集的預測結果取平均值用作第二層驗證集的一列特征表達。然后用CatBoost 和LightGBM 重復上面步驟。最后,在第二層中用隨機森林進行訓練預測,相當于對第一層的模型做了融合。
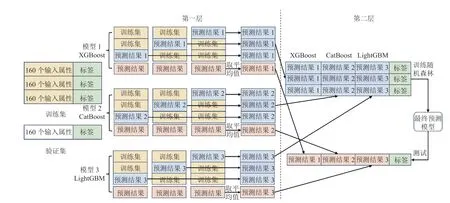
圖2 融合預測算法工作流程
本文融合模型交叉驗證結果如表5 所示,其中準確性ACC 為92.8%,MCC 達到85.5%,OPM 值為79.8%。

表5 融合模型10 重交叉驗證結果
3.4 盲測結果及與其他工具比較
以剩余的10%數據作為盲測集,使用構建好的融合模型進行預測。其結果如表6 第二列所示,本文融合模型準確性ACC 達到93.1%,MCC 為86.1%,OPM 為80.6%。
本文融合模型在盲測集上的ROC 曲線如圖3所示,曲線下面積(AUC)為0.921。
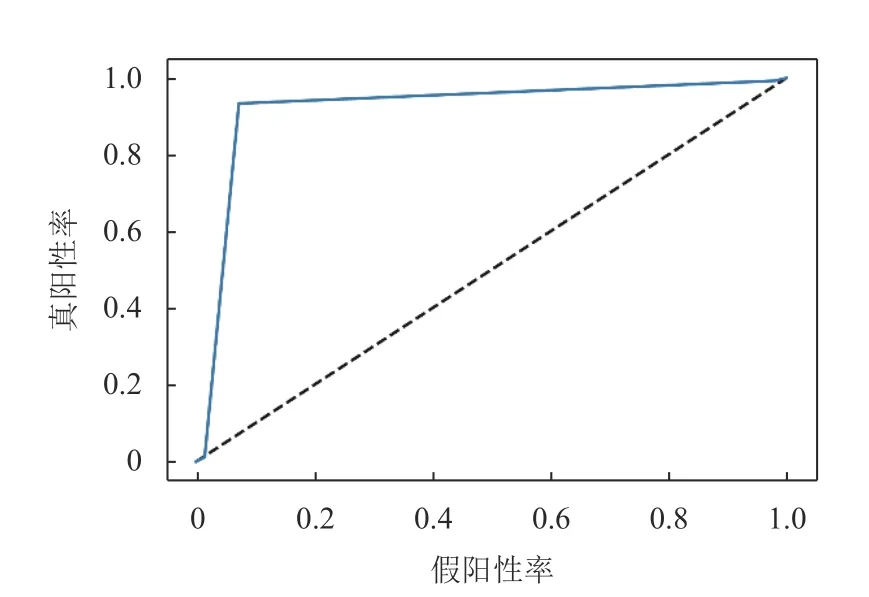
圖3 盲測集ROC 曲線
本文還在該盲測集上比較了幾種常見的預測工具性能,包括Polyphen2[1]、CADD[2]、PON-P2[3]、DEOGEN2[4]、MetaSVM[5]、ClinPred[6]和PrimateAI[8]。其中CADD 閾值設為20 時,其性能取得最佳。
由表6 可見,Polyphen2 準確性較低,為75.0%。考慮到Polyphen2 訓練的數據量僅為2 萬條,限制了它的性能。CADD 準確性達到77.5%。該模型以36 個其他預測模型的得分作為特征,或許某些預測模型并不能有效提高氨基酸變異致病性的預測性能。定義覆蓋率為模型能夠預測的變異數/測試集總樣本數量。PON-P2 準確性最高,達到93.7%,但覆蓋率僅為54.2%,這是因為PON-P2 舍棄了置信度不高的變異預測。而本融合模型覆蓋率達到了100.0%。由于綜合了兩個預測模型,ClinPred 準確性較高,為91.1%,但覆蓋率也僅為76.5%。PrimateAI 是一個基于深度學習的預測模型,直接從序列中提取特征,準確性較低,僅為56.0%。可見生物特征在變異致病性預測中扮演著更為重要的角色。
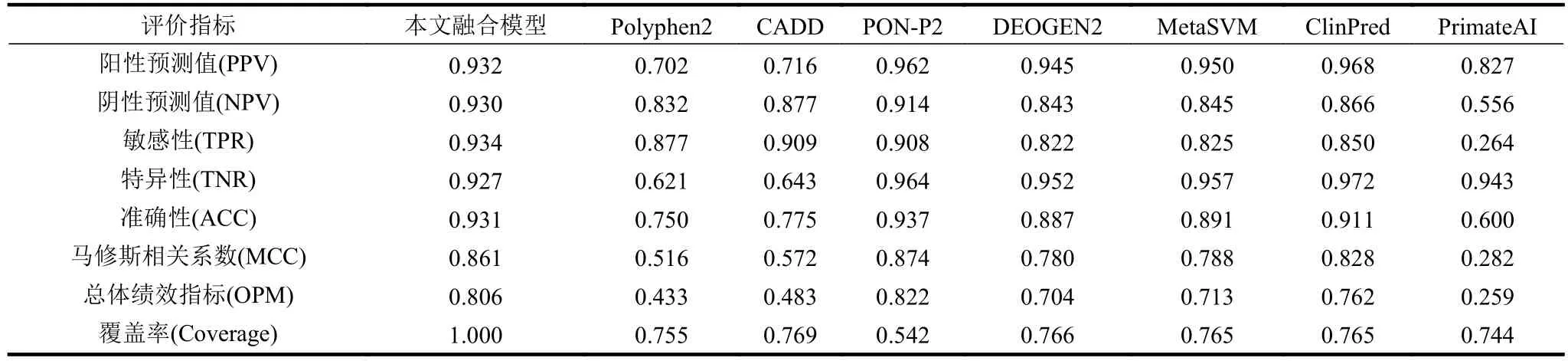
表6 與常用的預測模型在盲測集上的性能結果比較
4 結束語
氨基酸變異常常會對蛋白質的結構和功能造成影響,進而導致疾病。基于計算的方法作為一種預測氨基酸變異致病性的有效途徑,被研究者廣泛使用。為了提高預測準確性和泛化性,本文構建了一個新型融合模型。首先,收集到包含人類、動物和植物種群的氨基酸變異作為數據集。接著,提取有效生物特征,并用RFE 篩選出最優特征子集。然后,使用深度學習網絡提取特征,深度學習網絡由CNN 和Bi-LSTM 組成。將篩選完的生物特征和深度學習網絡提取的特征以拼接的方式融合,作為預測輸入。最后,構建一個基于XGBoost、CatBoost、LightGBM 和隨機森林的融合模型,得到最終的預測結果。融合模型的交叉驗證準確性ACC 達到92.8%,MCC 達到85.5%,OPM 值為79.8%。與其他工具相比,本文模型具有更高的準確性和泛化性。該模型工具可用于輔助臨床診斷和藥物設計,降低研發成本。
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
今日農業(2021年19期)2022-01-12 06:16:36
中老年保健(2021年11期)2021-08-22 03:15:44
中學生數理化(高中版.高考數學)(2021年1期)2021-03-19 08:28:38
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
現代出版(2020年3期)2020-06-20 07:10:34
數學物理學報(2020年2期)2020-06-02 11:29:24
瘋狂英語·新策略(2019年10期)2019-12-13 08:43:28
當代陜西(2019年10期)2019-06-03 10:12:04
數學小靈通·3-4年級(2017年9期)2017-10-13 08:10:54
