臨床研究樣本量的估計方法和常見錯誤
2022-02-14 02:26:08潘岳松金奧銘王夢星
中國卒中雜志 2022年1期
潘岳松,金奧銘,王夢星
近年來,隨著我國對臨床研究的重視,國內腦血管病領域的相關研究迅速發展。樣本量估計是臨床研究設計中的核心問題之一,隨著臨床研究的發展,也越來越受到關注和重視。最近的一項統計顯示,N Engl J Med發表的文章中,使用樣本量計算/統計功效分析的在1978-1979年只有39%,而在2015年增長到了62%,是目前該雜志發表文章中使用頻率最高的統計學方法[1]。樣本量的正確估算直接關系到臨床研究的成敗,也一直是臨床研究者較為關注且難以把握的關鍵點之一。如果樣本量太小,研究結果的可重復性及代表性就欠佳,可能得出假陰性或假陽性的結論;如果樣本量過大,研究所需的經費和資源就越多,項目執行的難度也越大,而且試驗過程中對研究對象可能的潛在傷害越大,還存在倫理問題。那么,臨床研究設計時如何確定合適的樣本量呢?本文對決定樣本量估計的要素、常用的腦血管病臨床研究樣本量計算方法、樣本量估計中常見的問題等方面進行闡述,以促進腦血管病領域的研究者更好地掌握臨床研究樣本量估算的方法。
1 決定樣本量估計的要素
1.1 研究目的、研究設計和主要觀察指標的資料類型 臨床研究的樣本量估計首先需考慮研究目的和研究設計類型,根據不同的研究類型來選擇對應的估計方法[2]。首先應明確研究的目的是分析疾病發病或預后的危險因素,是驗證某項干預措施的有效性和安全性,還是評估某項新技術診斷疾病的準確性。研究目的不同,對應的樣本量估計的考慮因素也不同。臨床研究設計類型是屬于病例對照研究,還是隊列研究、隨機對照試驗?隨機對照試驗是優效性設計,還是非劣效性設計?暴露因素/干預措施分組是兩組還是多組?是否匹配設計?不同的臨床研究設計類型對應的樣本量估計需考慮的因素和計算公式各不相同。在估計樣本量之前,首先要明確和充分理解研究的目的和研究設計的類型。
樣本量估計需考慮的另一個重要因素是主要觀察指標的資料類型。主要觀察指標的資料類型一般可以分為定性指標和定量指標兩種,對應的樣本量估計方法各不相同。腦血管病臨床研究中常用于評估二級預防效果的結局指標如卒中復發、聯合血管事件發生、缺血性卒中復發等;常用于溶栓、機械取栓、神經保護等臨床試驗的結局指標如功能預后良好(mRS 0~1分或mRS 0~2分);Ⅱ期臨床試驗常用的神經功能惡化/好轉等指標均為二分類定性指標。對主要觀察指標為二分類定性指標的臨床研究,樣本量估計主要采用率的比較計算公式,用到的主要參數為率。常用于卒中后認知功能障礙、腦小血管病預后研究的認知功能評分等指標為定量指標,對主要觀察指標是定量指標的臨床研究,樣本量估計主要采用均數比較的計算公式,用到的主要參數為觀察指標的均數和標準差。
1.2 效應值 暴露組或干預組間的主要觀察指標的預估效應值大小是直接決定臨床研究所需樣本量最主要的因素之一。對于兩組比較,以Δ表示兩組總體參數(均數或率)的差值或比值。常用的效應值包括組間MD、RD、RR、HR、OR等。一般來說,兩組的預估效應值越大,如兩組MD或RD越大,RR、HR或OR越偏離1,所需的樣本量越小。當臨床研究是優效性試驗或非劣效性試驗設計時,還需結合比較優效性界值或非劣效性界值來確定樣本量。
1.3 變異度 一般用方差或標準差反映組間觀察指標的總變異程度。兩組定量指標(均數)的比較,其方差可通過兩組樣本方差估計。兩組定性指標(率)的比較,其方差可通過兩組樣本率估計。一般情況下,變異度,即方差越大,所需樣本量越大。事件率越接近0.5,所需樣本量越小。
1.4 檢驗水準 檢驗水準α,即Ⅰ型錯誤的概率,是指錯誤地拒絕了實際成立的原假設H0,錯誤地判定為有差異的概率大小。α越小,所需樣本量越大。α的取值常為雙側0.05或0.1,優效或非劣效試驗設計中常取值為單側0.025。
當需要多重檢驗時,如設置了多個主要療效指標、擬進行多組間兩兩比較或在試驗過程中設計了期中分析,需進行多次比較分析的情況下,則會使Ⅰ型錯誤增加,需對α進行校正。在這一過程中要進行多次重復顯著性檢驗,每進行一次檢驗都將增加Ⅰ型錯誤的概率,從而使總的顯著性水平α上升。如,以檢驗水準α=0.05,重復進行10次檢驗為例,發生Ⅰ型錯誤的總概率將上升到0.19。常用的調整檢驗水準α的方法,如Bonferroni法,調整后的α’=α/k(k為統計檢驗的次數),如總共需進行3次檢驗,則α’=0.05/3,為0.0167。成組序貫研究設計包含了期中分析,為了使總體顯著性水平維持常數α,必須調整每一次分析的顯著性水平,常用的調整α水平的方法包括Pocock法、O’Brien-Fleming法和Peto法等[3]。
1.5 把握度 把握度,即檢驗效能,是指所研究對象總體間確有差異時,按檢驗水準α能夠發現此差異的概率。把握度=1-β,其中β為Ⅱ型錯誤的概率,因此指定了β水平也就等于指定了把握度水平。把握度越大,所需樣本量越大,通常將其定為0.80或0.90。一般建議臨床試驗把握度定為0.90。
1.6 其他因素 除了上述主要因素外,其他因素如兩組例數的分配比例、優效性與非劣效性界值、不應答或失訪率等均可影響樣本量的估計[4]。一般而言,兩組比較時取相等的樣本含量,此時總的樣本含量最少,且可達到最高的統計效能,因此經常使用的是各組等樣本含量設計。但是,由于某些實際原因,有時可能需要進行各組不等樣本含量設計,在進行樣本量估計時也應予以考慮。通過樣本量估算公式計算得到樣本量后,一般要考慮不應答或失訪的影響,增加相應的樣本量,以確保實際收集的有效病例數能足夠達到統計要求。如考慮失訪的影響,可將最后的樣本量定為N’=N/(1-失訪率)。
2 樣本量估計的思路
不同的臨床研究設計樣本量估計方法也不同,樣本量估計的思路非常重要(表1)。與臨床研究樣本量估計相關的最核心的參數是“兩組主要觀察指標的預估值”。在根據樣本量估計公式計算之前,需要先明確3個問題:①研究的目的是什么?采用什么樣的設計?②研究的主要觀察指標是什么?③主要觀察指標有預估值嗎?其中,研究目的、研究設計和主要結局指標在研究方案設計時應該就已經確定了。而主要觀察指標的預估值,則可通過預試驗或總結前期數據、查閱文獻及結合專家意見,由臨床專家和統計學專家聯合確定。確定了上述3個問題的答案,就確定了研究樣本量估計時需要用到的參數及參數的大小,之后將數據代入樣本量計算公式,采用軟件計算即可。常用的PASS、SAS、Stata等統計軟件以及一些公眾網站和微信小程序都可以方便地實現樣本量的計算[5]。

表1 樣本量估計的思路
3 常見的臨床研究設計樣本量估計方法
3.1 臨床登記隊列樣本量估計 在腦血管病臨床研究中,最常見的研究設計類型是臨床專病隊列,用于探討基線預后影響因素與患者預后的關系。根據危險因素的有無或高低可分為兩組,進而比較兩組卒中復發率的差異或檢驗暴露因素與患者預后的關系。兩樣本率比較的樣本量估計可采用公式①進行計算。其中p1與p2分別代表兩組的率,p=(p1+p2)/2。有時實際得到的是對照組的率p1和效應值RR或OR值,可通過公式p2=RR×p1或公式p2=(OR×p1)/[1+p1×(OR-1)]進行轉化。Z1-α/2和Z1-β分別為α和1-β對應的標準正態分布臨界值。實際工作中,可進一步考慮兩組不等比例、混雜因素的影響等對樣本量做進一步調整,擴大樣本量。

示例:某研究者擬進行一項臨床登記研究探討基線糖尿病狀態與卒中患者1年卒中復發的關系。前期資料顯示糖尿病的RR值為1.3,無糖尿病人群的一年卒中復發率為8%,取α=雙側0.05,β=0.20,考慮失訪率為10%,計算得所需樣本量為2529例/組。
3.2 優效性臨床試驗兩樣本率比較 優效性臨床試驗的目的是評價試驗干預措施是否優于對照措施(陽性或安慰劑對照)。如雙聯抗血小板治療對比單聯抗血小板治療的試驗、機械取栓對比傳統治療的試驗均多采用這種研究設計[6-7]。兩樣本率比較優效性臨床試驗的樣本量估計可采用公式②進行計算。其中p1與p2分別代表兩組率,p=(p1+p2)/2,Δ為優效性界值。Δ一般取具有臨床意義的最小值,由臨床專家與統計學專家協商確定。當Δ=0時,優效性檢驗相當于兩樣本率比較的樣本量計算方法。
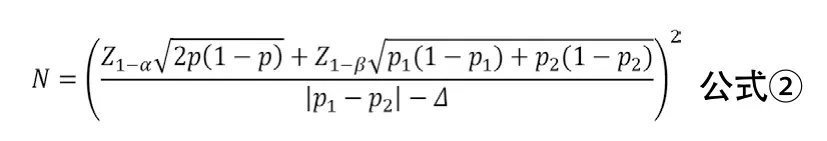
示例:某試驗擬比較阿司匹林聯合氯吡格雷雙聯抗血小板治療與單用阿斯匹林治療對預防卒中復發的療效,根據前期的文獻報告,預設阿司匹林聯合氯吡格雷雙聯抗血小板治療組3個月卒中復發率為8%,單用阿斯匹林治療組3個月卒中復發率為10%,優效性界值Δ=0,α=0.025,β=0.10,考慮失訪率為5%,計算得所需樣本量為4528例/組。
3.3 非劣效性臨床試驗兩樣本率比較 非劣效性臨床試驗的目的是評價試驗干預措施在臨床意義上不差于(非劣于)對照措施(通常為已上市的有效藥物或標準治療方案),目的是探索新的治療選擇。這種情況常用于原有上市藥物的療效較好,試驗藥的療效超過標準治療措施的可能性較小,預計研究藥物的療效與原有上市藥物相當,但研究藥物可能具有其他特點,在其他方面可能有優勢,如更好的安全性、使用更方便或依從性更好等。隨著藥物研發的深入,近年來采用非劣效性設計的試驗越來越普遍。腦血管病臨床研究中如比較低劑量對比標準劑量阿替普酶溶栓、直接取栓對比橋接取栓、取栓前替萘普酶對比阿替普酶溶栓等試驗多采用這種設計[8-10]。
兩樣本率比較的非劣效性臨床試驗的樣本量估計可采用公式③進行計算。其中p1與p2分別代表兩組率,p=(p1+p2)/2,Δ為非劣效性界值。非劣效性臨床試驗通常設兩組的率相等。Δ一般由臨床專家與統計學專家根據既往研究證據結合臨床意義共同確定,并最終由臨床專家確認。統計上可采用兩步法估算,先估計出陽性對照藥物相對于安慰劑為對照的絕對療效M1,一般取小于陽性對照與安慰劑效應之差的95%CI下限(高優指標)[11]。臨床可接受的非劣效性界值M2一般通過M2=f×M1計算確定,建議非劣效設計中取f=0.5。在沒有歷史數據可依據時,Δ的確定也可根據目標值法取值為陽性對照藥物療效的10%~15%。
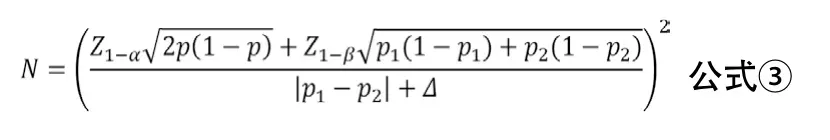
示例:某試驗采用非劣效性試驗設計,擬檢驗直接取栓治療患者3個月預后良好的比例不劣于橋接取栓治療的患者。根據前期數據和文獻報告,預設兩組患者3個月預后良好率為60%,非劣效性界值Δ=5%,α=0.025,β=0.10,考慮失訪率為5%,計算得所需樣本量為2124例/組。
以上為腦血管病臨床研究中常見的樣本量估計方法,特殊類型研究,如整群隨機對照臨床試驗、適應性設計臨床試驗、單組目標值試驗、診斷性試驗等的樣本量計算,以及兩樣本均數比較的樣本量估計方法,可參閱相關文獻,因為篇幅的限制,本文不作詳述。
4 樣本量估計的常見錯誤
4.1 沒經過計算直接確定樣本量 部分研究者在撰寫臨床研究方案時,未經過計算就直接確定樣本量(如100例或200例)。這種確定樣本量的做法可能導致樣本量不足,達不到統計學檢驗的要求,得不到預期的研究結果。不過,對于因實際情況限制無法入組過多的研究,或研究本身的目的是為了進行預試驗探索方案的可行性、初步探索干預措施的療效和安全性,可不按照樣本量估計的例數入組,但應對預試驗的研究目的給予明確說明。
4.2 樣本量估計方法與研究設計和主要觀察指標不對應 在樣本量估計過程中,最常見的錯誤之一是樣本量估計方法與研究設計和主要觀察指標不對應。如:研究的目的是采用隊列研究設計驗證基線時某指標升高與卒中預后的關系,但樣本量估計時卻依據1年卒中復發率,采用率的橫斷面調查公式的方法進行樣本量估計;或者,臨床試驗中設置的主要觀察指標為3個月預后良好的比例,但因為缺少前期數據,主要觀察指標無法預估,在樣本量估計時采用了3個月卒中復發率作為計算的依據。樣本量的估計方法應與研究目的、研究設計和主要觀察指標相對應,否則無法得到預期的效果。
4.3 參數設置不合理 樣本量估計的另一個常見錯誤是樣本量估算的參數設置缺乏依據,或參數設置不合理、不符合臨床實際情況。如:為節省樣本量有意夸大事件率或預期的效應值,或設置的脫落率和失訪率過高,設置的參數明顯不符合臨床的實際情況。上述情況都可能導致樣本量估計不準確,從而使研究達不到預期的研究效度。
總之,在臨床研究的過程中,樣本量既不是越大越好,也不是越小越好。合理的樣本量是臨床研究科學設計的重要環節,與研究設計的其他環節密切相關,估計過程應充分理解和考慮研究目的、研究設計和主要觀察指標的資料類型。樣本量需要臨床專家和統計學專家合作討論確定,選擇正確的計算方法和公式,合理設置參數,并進行科學的計算才能保證其準確無誤。
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
現代裝飾(2020年7期)2020-07-27 01:27:42
流行色(2020年1期)2020-04-28 11:16:38
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
藝術啟蒙(2018年7期)2018-08-23 09:14:18
海峽姐妹(2017年7期)2017-07-31 19:08:17
汽車工程學報(2017年2期)2017-07-05 08:13:02
