冗余分析在微生物生態學研究中的應用
2022-02-16 23:20:25任玉連,董醇波,邵秋雨,張芝元,梁宗琦,韓燕峰
山地農業生物學報 2022年1期
關鍵詞:相關性
任玉連,董醇波,邵秋雨,張芝元,梁宗琦,韓燕峰
摘要:冗余分析(Redundancy analysis,RDA)是多元回歸模型的延伸,其用排序的方法對環境變量與物種間關系進行間接梯度分析,這一方法在經典生態學研究中已取得大量研究成果。本文闡述了冗余分析的概念,并在CANOCO軟件RDA分析操作步驟的基礎上,綜述了近年冗余分析在微生物群落多樣性、碳源代謝、核心微生物組、微生物譜系地理、病原菌耐藥性等微生物生態學中的重要應用,以期為微生物生態學研究推薦和擴展這一簡潔、快速的研究分析方法。
關鍵詞:冗余分析;間接梯度分析;環境變量;相關性
中圖分類號:Q938.1文獻標識碼:A
文章編號:1008-0457(2022)01-0041-008國際DOI編碼:10.15958/j.cnki.sdnyswxb.2022.01.006
Application of Redundancy Analysis in Microbial Ecology
Ren Yulian,Dong Chunbo,Shao Qiuyu,Zhang Zhiyuan,Liang Zongqi,Han Yanfeng*
(Institute of Fungus Resources,Department of Ecology,College of Life Sciences,Guizhou University,Guiyang,Guizhou 550025,China)
Abstract:Redundancy analysis (RDA) is an extension of multiple regression model.The relationship between environmental variables and species is indirect gradient analysis by using the ranking method.This method has achieved a lot of research results in classical ecological research.Based on the operational steps of RDA analysis of CANOCO software,the concept of redundancy analysis was introduced,and the important application of redundancy analysis in microbial community diversity,carbon metabolism,core microbiome,biogeographic distribution of microbial genealogy,and pathogen resistance were reviewed in recent years.We aimed to recommend and expand this simple and rapid analysis method for studying the microbial ecology.
Keywords:redundancy analysis;indirect gradient analysis;environment variable;correlation
冗余分析(Redundancy analysis,RDA)是多元回歸模型的延伸,其用排序的方法闡述群落生境或其中某一個生態因子隨樣地生境的變化。由于其能快速獲得解釋變量與響應變量間的關系,近年來在生態學領域的研究中受到越來越多的重視[1]。由生物或非生物介導的地上或地下等復雜的生態過程中,影響因子絕不是單一的,其作用也不可能獨立。冗余分析經過對特征值進行一系列分解篩選,可實現有效簡化目標變量個數的目的,進而將物種與環境因子的關系直觀地體現在同一坐標軸上,最終獲得解釋變量與響應變量之間的關系,獲得一個或多個主導因素[2]。此外,冗余分析還能獨立保持各個解釋變量對響應變量的貢獻率,同時對具體指標解釋能力大小及排序可靠性進行定量描述,以及具有識別環境變量組合的主要選擇性梯度的優勢[3]。因此,使用冗余分析能快速有效地解決應用研究中的多因子影響貢獻率排序問題。
目前,許多學者使用CANOCO軟件進行冗余分析。該軟件在解釋大尺度宏觀生物群落結構、動植物區系與環境之間的關系,檢驗假設沖擊對環境和其生物群落所造成影響的程度,以及在分析不同的生態系統和生態毒理學等領域具有廣泛的運用[4]。自1985年CANOCO 1.0發布以來,已發布了6個版本,現廣泛使用的是CANOCO 5[5]。該軟件包含了經典RDA冗余分析、梯度分析、分類、多元回歸、相似性度量和實驗設計等排序方法。由于其操作簡單、功能齊全,是生態學及相關領域多元數據排序分析的流行軟件之一[2]。
山地農業生物學報2022年第1期任玉連,等:冗余分析在微生物生態學研究中的應用冗余分析的應用已從宏觀生態分析,如與水生動植物、鳥類、植被、土地利用類型及景觀格局分布的關鍵環境因子篩選,逐漸被運用到微生物生態學及其與人類相關疾病等領域[6]。在微觀土壤微生物生態學研究中,冗余分析被用來進行微生物群落結構、組成、多樣性及功能等方面主控因素的提取。在這類冗余分析時,一方面需要考慮單個因素對微生物的影響;同時,也需要分析多個因素對微生物的影響程度[7]。
冗余分析中的數據選擇是靈活分析不同解釋變量與響應變量關系的核心。在微生物生態學研究中,多數學者將微生物群落結構及多樣性作為一組變量,而把環境因子作為另一組變量進行冗余分析,這樣既能很好地揭示環境因素對微生物群落結構及多樣性整體的影響,也能很好地揭示環境因子對各物種的影響貢獻大小。如Yang等[8]為弄清土壤微生物群落結構的關鍵作用和潛在機制,基于分類距離和系統距離對細菌群落進行冗余分析。Cao等[9]基于Biolog生態平板數據,以確定土壤生化特性與細菌群落組成和功能多樣性間的關系,將土壤環境變量作為解釋變量,而將不同處理下微生物代謝功能頻率分布變化,作為響應變量進行冗余分析。此外,Wang等[10]則將針葉林和闊葉林的凋落物特征、土壤性質作為解釋變量,土壤微生物群落則為響應變量,進行冗余分析后,快速獲得了土壤特性、凋落物性質對微生物群落結構總變異的解釋量排序。因此,在進行冗余分析時,需針對不同的研究目的,謹慎選取解釋變量與響應變量是正確獲得冗余分析結果的關鍵。
1冗余分析概念
冗余分析(Redundancy analysis,RDA)是一種回歸分析結合主成分分析的排序方法,能對多個解釋變量(x1…xn)的多個響應變量(y1…yn)進行回歸分析,將解釋和響應兩組變量的數據集通過雙標圖顯示二者間的相關性變異程度[4]。如在一元線性回歸分析中,每一個物種的多度y通過對應的解釋變量x進行回歸分析:
yik=ak+bkxi+error(1)
yik是樣方i物種k的多度,xi是樣方i的已知解釋變量。將物種的多度和解釋變量定義為一種線性關系,ak和bk分別是這一直線的截距和斜率。通過環境變量的加權總和約束x。以2個環境變量為例:
xi=c1zi1+c2zi2(2)
可獲得最優權重c1和c2,其權重值為典型相關系數。將(2)代入(1)中可得:
yik=ak+bkc1zi1+bkc2zi2+error(3)
通過物種數據yik和環境數據zi1獲得模型中的物種參數ak和bk(1,…,m)以及權重值c1和c2,即
d1k=bkc1,d2k=bkc2(4)
將模型(3)改為:
yik=ak+b1kzi1+b2kzi2+error(5)
綜上,冗余分析是一個對所有物種同時進行多元回歸的模型,既是一種約束性的主成分分析,也是一種約束性的多元多重回歸。
此外,冗余分析可將一維模型推廣到多維。以兩維模型為例:
yik=ak+bk1xi1+bk2xi2+error(6)
bks為第k個物種的得分,xis是第i個樣方在第s(s=1,2)排序軸上的得分。樣方得分通過(7)進行約束:
xis=c1szi1+c2szi2(7)
cjs為第j個環境變量在s排序軸上的典型相關系數。將(7)代入(6)可得回歸系數受約束后的多元回歸模型。以djk代表回歸系數為例:
djk=bk1cj1+bk2cj2 (8)
該模型是回歸雙標圖的基礎,通過(6)可看出樣方得分xis和物種得分bks共同形成物種擬合值的雙標圖[11]。
2基于CANOCO 4.5進行冗余分析操作CANOCO 4.5軟件主要包括Canoco for Windows、WcanoIMP、CanoDraw、CanoMerge和PrCoord五大模塊。其中,Canoco for Windows用來指定需分析的數據和排列模型;WcanoIMP將Excel等形式的數據轉化為CANOCO能識別的*.dta形式;CanoDraw進行排序圖的生成,以及生成不同類型的等值線和回歸模型;CanoMerge是合并CANOCO能識別的*.dta類型數據文件,可將數據文件以帶制表分隔符的文本形式輸出,具有濾掉低頻率物種的功能;PrCoord對特定數據集進行主坐標分析以及冗余分析[2,4]。
2.1軟件安裝
下載國際通用軟件包CANOCO 4.5,解壓并按照提示安裝程序updt_456.rar安裝軟件。
2.2原始數據
輸入數據時應遵循樣方名和變量名命名規則,一般來說字符不得超過8個;建議使用簡單的數字、字母、點、連接符和空格結合進行命名;在Excel中將物種變量和環境變量按照樣方名稱和變量名稱命名格式輸入需要分析的數據。確保每一行為一個樣本,每一列為一個物種(或變量)。如果物種變量和環境變量之間有數量級差別,則需要對數據進行數據標準化處理(如對數轉換、ZScoring等)。
2.3數據轉換
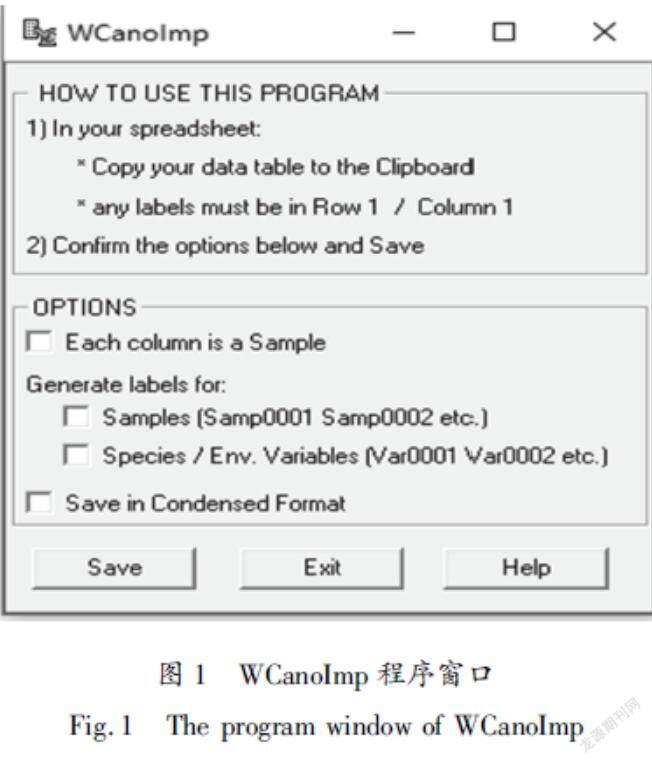
CANOCO 4.5版本很好地解決了數據文件過于復雜和格式問題。即用WCanolomp將兩組數據轉換為CANOCO能識別的格式文件。打開已備好的原始數據,分物種變量和環境變量拷貝后,訪問WCanoImp.exe程序。如圖1對話框所示,確定所選選項正確后,單擊保存按鈕繼續,指定要生成的文件夾和存儲位置,用于正在生成的數據集。同時,WCanoImp程序運行的另一個對話框會顯示成功創建。此時,Excel數據已轉化為CANOCO可識別數據(后綴以*.dta命名,如“spe.dta”“env.dta”)。
2.4排序方法的選擇
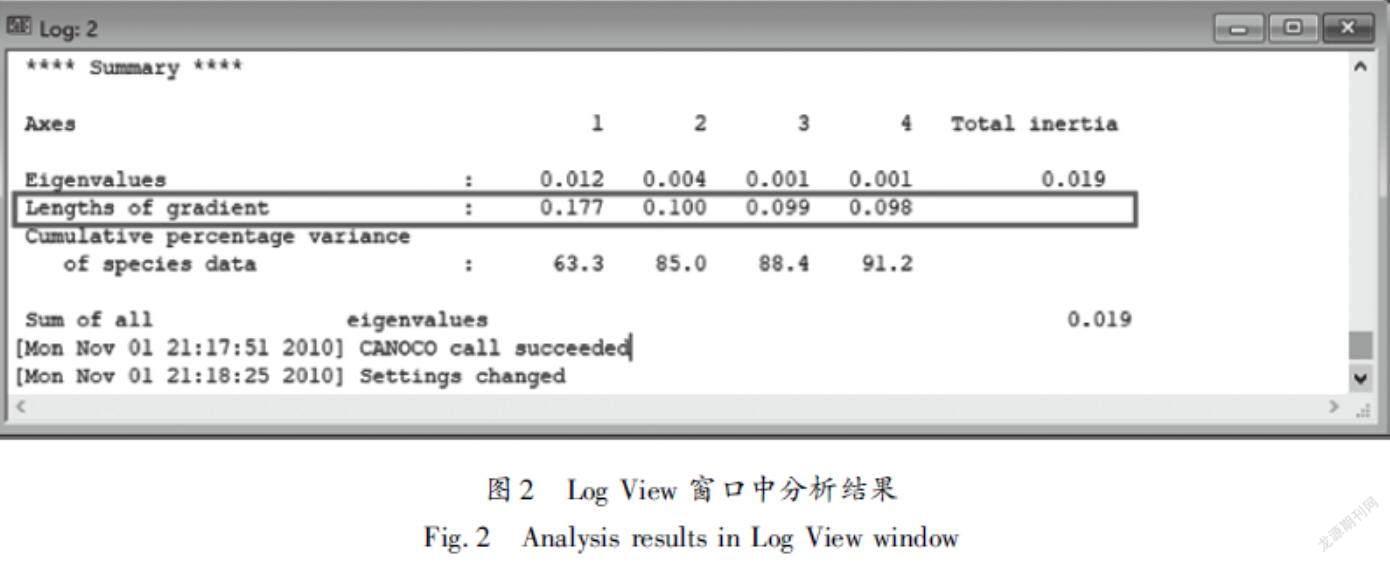
CANOCO 4.5版本能快速進行去趨勢分析(Detrended correspondence analysis,DCA),它是判別使用RDA或典范對應分析(Canonical correspondence analysis,CCA,也是一種分析生物群落與環境因子間相互關系的一種方法)的判斷依據。利用Canoco for windows>New project>Available Data>Next>Data files>Browse指定要分析數據文件>下一步>DCA>Finish Options>點擊“Project View”窗口的“Analysis”>分析結果即可在“Log View”窗口中查看。在得到4個排序軸的梯度長度后,根據DCA分析結果中的Lengths of gradient數值大小判斷選取RDA或CCA方法。梯度長度<3時,使用RDA線性響應模型(圖2);梯度長度介于3~4之間,RDA與CCA分析均可;梯度長度>4時,選擇CCA單峰響應模型進行分析[12]。
2.5RDA模型建立
在CANOCO 4.5菜單欄上依次點選:File>New project>Available Data界面,選擇“Species and environment data available”> Next,將上述“spe.dta和env.dta”文件對應添加到“Data”Files”界面的前兩列中,最后一列是輸出“*.sol”文件>下一步> RDA>Scaling Linear Methods>Symmetric>Divide by standard deviation>Finish Options>Canoco Project,點擊“Project View”窗口中“Analysis”> 保存生成的“*.Log”文件,可在“Log View”窗口中查看分析結果。在RDA分析中,需要對環境變量選擇手動篩選(Manual selection),在Project>Analysis中彈出的對話框中,依次檢驗環境變量的解釋度(Test variable),根據P值大小,選擇需要的變量。
2.6RDA模型作圖
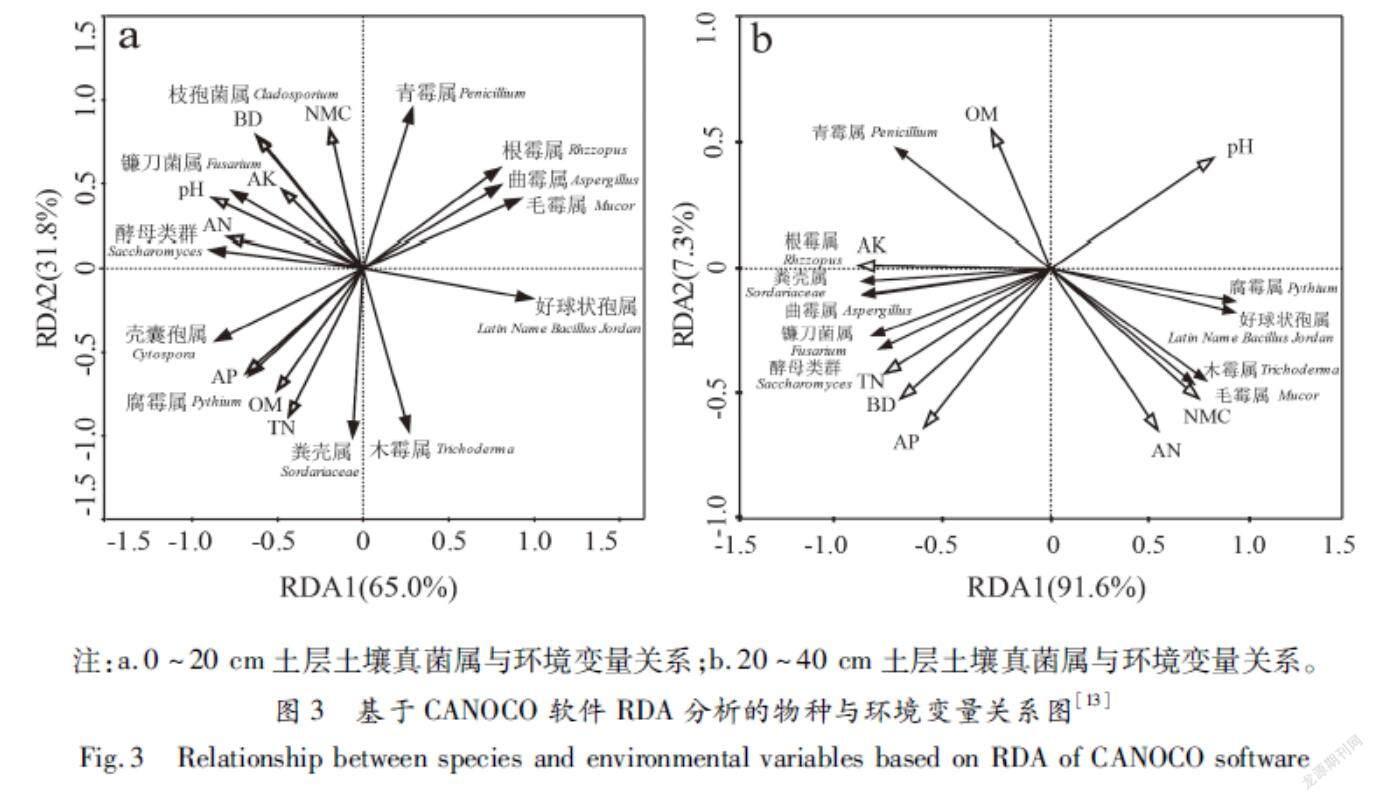
從上一步驟中的Project view >激活CanoDraw.exe>Create>Biplots and Joint Plots>Species and env.Variables / Species and samples / Samples and env.variables即可對物種以及環境變量/物種以及樣品/樣品以及環境變量作圖。最后將生成的RDA圖經線條、顏色、字體等調整后保存并輸出。如圖3所示,排序軸以土壤真菌被理化因子的線性組合解釋量的多少先后出現(排序軸1,2,3和4……n),前四軸通常會占到解釋量的較大部分,因此在排序圖中一般只選取前四軸中的兩個進行制圖。
2.7冗余分析排序圖的解釋
圖3為真菌屬分類水平與土壤環境變量間的冗余分析結果。其中,黑色實心箭頭代表各類真菌屬;黑色空心箭頭代表理化因子;黑色實心箭頭連線與黑色空心箭頭連線之間的夾角代表某類真菌屬與某理化因子之間的相關性,用夾角的余弦值表示;即箭頭連線與排序軸的夾角表示真菌屬與理化因子間的相關性。夾角接近90°為接近正交,表明真菌屬與理化因子之間的相關性很小,二者間幾乎不存在影響(圖3b:OM與鐮刀菌屬)。夾角小于90°為銳角,表明真菌屬與理化因子之間存在正相關(圖3b:AP與曲霉屬);銳角角度越小,則正相關性越大(圖3a:AP與腐霉屬)。夾角大于90°為鈍角,表明真菌屬與理化因子之間存在負相關(圖3b:pH與糞殼屬);鈍角角度越大,則負相關性越大(圖3a:TN與青霉屬)[14]。黑色空心箭頭所處的象限代表理化因子和排序軸的正負相關性;黑色空心箭頭連線在排序軸上投影的長短表示某個理化因子與排序軸之間相關性大小,投影長度越長,則相關性越大。圖3冗余分析顯示,土壤不同理化因子影響其不同真菌類群的生存繁殖。0~20 cm與20~40 cm土壤中,第Ⅰ軸和第Ⅱ軸真菌群落特征—土壤理化因子關系累計解釋量分別高達65%和91%以上(圖3 a、b)。0~20 cm土層,有機質、全氮和速效磷等理化因子對50%以上的菌群具有促進或抑制作用,是影響真菌群落結構的重要因子,而20~40 cm土層影響其真菌群落結構的是速效鉀、全氮、容重、自然含水率和pH等理化因子。說明土壤理化因子在決定土壤真菌群落結構分布中起著重要作用[13]。
3 冗余分析在微生物生態學研究中的應用冗余分析作為生物與環境變量關系的一種測度和類型[7],與回歸分析、相關分析、主成分分析、結構方程模型以及可視化網絡等分析方法目的相同,均是尋找解釋變量與響應變量之間的關系。由于冗余分析的簡潔、快速和可視化的特點,在微生物生態學研究領域中,已越來越多的受到研究者的關注。
3.1 微生物群落組成及多樣性與環境因子間關系分析土壤細菌、真菌和放線菌等群落結構組成及多樣性變化可敏感的指示土壤環境條件變化 [13,15]。因此,不同的研究目標所選取的解釋變量和響應變量差異巨大。基于目水平的豐富度指數(Chao和ACE)及多樣性指數(Simpson和Shannon)為響應變量,以速效氮、有效磷、速效鉀、全氮、有機質和pH為解釋變量,結果表明,所有環境因子解釋了細菌和古菌菌群的變化,貢獻大小依次為pH>速效鉀>有效磷>有機質,其中pH對土壤細菌和古菌群落結構的影響最大,解釋了66.5%的變化;其他環境因子則共解釋了細菌與古菌群落33.5%的變化[16]。基于門水平16S rRNAs功能基因豐度,Shu等[17]使用了更多的環境參數,如pH、NH+4N、NO-2N、COD(化學需氧量)與群落結構間的關系發現,厭氧氨氧化細菌與NH+4N、NO2N去除率呈正相關。Cui等[18]基于普通細菌、革蘭氏陽性菌、AM真菌、厭氧菌和放線菌作為響應變量;C:N、C:P、N:P、pH、Fe、Mn、EC(電導率)為解釋變量;分析結果顯示,細菌、革蘭氏陽性菌、AM真菌、厭氧菌和放線菌與碳氮比呈顯著負相關,與碳磷比、氮磷比無顯著相關性。真菌和放線菌與鐵和鈣呈正相關,與酸堿度呈負相關。近年來,還有不少研究者以SOC、TN、TP、TS等為響應變量,以其他化學成分及計量比BD、EC、水分、SOM、CEC(陽離子交換量)、K+、Ca2+、Mg2+、N-源、C/P、C/S、C/N等,以及土壤類型為解釋變量[19],在揭示環境非生物因素對土壤及菌根微生物群落組成和結構上影響度獲得了新的結果。
3.2碳源代謝分析
微生物代謝多樣性能夠反映微生物群落的生態特征[20]。由于土壤微生物的碳源代謝能力受土壤性質、植物多樣性、凋落物組成等因素影響,但是對于不同生境中的研究,不同學者選取的解釋變量和響應變量具有一定差異。如王蕓等[21]基于含水量、沙礫和容重為解釋變量,以微生物碳源代謝為響應變量的分析結果顯示,含水量、沙礫和容重顯著影響土壤微生物碳源代謝功能,分別能解釋105%、88%和72 %的變異。土壤pH值被用來解釋微生物代謝能力的重要變量之一,因為隨著土壤pH的減小,土壤代謝強度顯著增加[22]。此外,電導率、全氮和堿解氮能顯著促進微生物的碳源代謝活性,土壤鹽度則顯著抑制了土壤微生物的碳源代謝活性,也被選作冗余分析時的解釋變量,特別在不同經營模式林分土壤微生物的碳源利用研究中被廣泛使用[20]。此外,由于土壤理化性質、植被多樣性和水熱條件等能影響土壤微生物代謝作用釋放分泌某些酶類,因此土壤酶活性被用來作為冗余分析中重要的響應變量之一[23]。如任玉連等[12]使用樣地微氣候(年均氣溫、年均降水量)、植被多樣性指數(Margalef豐富度指數、Shannon)樣性指數、Pielou均勻度指數及Simpson指數)和土壤理化性質(土壤密度、土壤溫度、含水量、pH、有機碳、全氮、水解性氮、全鉀、速效鉀、全磷、有效磷及C/N)與土壤酶活性(脲酶、蔗糖酶、酸性磷酸酶、多酚氧化酶、過氧化氫酶)進行冗余分析,最終獲得單一環境因子對土壤酶活性影響的重要性排序。曹聰等[24]認為使用冗余分析排序圖可以看出理化性質(溫度、水分、碳、氮和磷)對酶活性有不同程度的影響。因此,使用冗余分析可以快速獲得環境因子與微生物碳源代謝能力間的關系,但在選取解釋變量的過程中,應考慮生境條件及試驗條件的差異,盡可能獲得更為豐富解釋變量數據進行冗余分析。
3.3核心微生物組及功能分析
核心微生物組研究最早開始于人體的體表面和內部共棲、共生微生物組的部分成員,現已廣泛涉及動物、植物、土壤、水體以及廢水處理系統微生物組中有關的成員,它們是微生物組的基本組成及功能的關鍵部分[25]。由于核心微生物組在分類學上呈現的生物多樣性特性,與相關物種共存機制以及在群落功能中的作用相關,因此多數學者僅關注核心微生物組間的關系。目前主要基于分子技術用MetaCoMET、COREMIC、PhyloCore、BURRITO等分析工具綜合界定核心微生物組[26],但核心微生物組與環境變量間的關系較少被關注。Mukhtar等[27]在門水平上以古菌、細菌和真菌為響應變量;以頭孢氨芐、溫度和水力保留時間為解釋變量,分析揭示環境因素對細菌、真菌和古細菌群落結構的影響。此外,冗余分析也被用于尋找功能微生物(降解有機污染物的微生物菌群)中的中樞(hub)菌屬[28]。在揭示綠肥處理影響微生物相關功能基因的重要因素的冗余分析中,則以微生物功能基因相對豐度為響應變量,以碳、氮、磷、鐵、硫、pH以及植物激素為解釋變量[29]。此外,也有學者采用冗余分析對鹽脅迫下的核心微生物分類菌群進行研究[30]。
3.4微生物譜系地理
生態系統中碳、氮、磷等循環可能將生物地球化學模式與生理限制聯系起來,是微生物多樣性和營養限制的主要驅動力和關鍵預測因子[18,31]。由于不同物種的分布模式差異巨大,探索哪些因素是影響物種分布模式、遺傳多樣性和種群結構及群落擴散的關鍵因素尤為重要。因此,冗余分析也與其他分析工具相結合,從而實現不同的研究目的。如冗余分析與結構方程模型結合,用來揭示不同區域水體養分特征(鐵、錳、化學需氧量、營養鹽和酸堿度)對藻類群落影響的主要環境因子。運用冗余分析驗證緯度生物地理模式,并描述生物地理模式形成因素和關鍵分單元類群(Keystone taxa)[32]。通過冗余分析解釋低和高核酸含量細菌的豐度和細胞計數特征隨地理距離和環境變量(pH、溫度、鹽度、經度、緯度、海拔、溶解氧、電導率、葉綠素a)的變化,結果表明細胞計數特征和豐度與環境因素顯著相關,且pH是驅動低和高核酸含量細菌變化的主導因素[33]。這些結果顯示了地理距離和環境改變是影響微生物地理分布的主要因素。
3.5病原菌耐藥性的環境因素分析
2020年新冠肺炎疫情危機,是一場人與微生物病毒的無聲“爭斗”,這種微生物給人類的健康和生存帶來了巨大的威脅,生物安全問題已備受全球關注。微生物是導致人類傳染病流行的最重要因素之一 [3435]。近年來,人類在疾病的預防和治療方面取得了長足的進展,但現代社會廣譜抗生素的普遍使用已使許多菌株發生變異,導致耐藥性的產生,新微生物感染不斷發生。抗生素耐藥基因在環境中的擴散已成為日益嚴重的健康風險[36]。因此,基于冗余分析的便捷快速等優點,冗余分析可能在尋找影響微生物耐藥性環境因素方面具有廣闊的應用前景。如基于抗生素、金屬、鎂元素和細菌為環境變量,以精氨酸為響應變量,冗余分析發現,抗生素、金屬、鎂元素和細菌分別解釋了微生物群落中精氨酸變異的07%、57%、124%和219%的貢獻率[36]。此外,Guo等 [37]以OTC(土霉素)和Cd(鎘)等為解釋變量,以抗生素耐性基因和微生群落為響應變量進行冗余分析,獲得OTC和Cd等因素對微生物抗生素耐藥性基因變化的解釋率。Zhang等[38]基于冗余分析揭示添加鎘對土霉素污染土壤中精氨酸轉運、微生物群落和人體病原菌的影響。由于影響人體微生物菌群的因素眾多,因此,在開展這一領域的研究時,獲得的解釋變量越多,采用冗余分析能快速獲得不同解釋變量的貢獻率,可能其實際運用價值更高。
4展望
冗余分析結合環境矩陣和物種矩陣可直觀、明確、有效地揭示環境因子與物種間相關程度的大小,是一種經典、成熟的有效方法,在傳統生態學各領域研究中具有較為突出的優勢。由于這種分析方法操作簡單、快捷,在處理微生物組產生的巨量數據面前,能較快地找到環境與物種間的復雜關系并對變量之間的關系作出直觀的生態解釋,凸顯了它的優點和優勢。故已在不同生態系統中微生物組的組成、結構、功能、生物多樣性及碳源代謝方面皆展現了廣闊的應用前景。今后,冗余分析解析方法主要從以下幾個方向發展。
4.1解釋變量的高度選擇
微生物生態研究中使用冗余分析時,解釋變量的指標選取,成為影響研究結論的核心因素。由于環境因素復雜多樣、群體大、且各群落間存在許多促進和抑制的關聯,為能在眾多解釋變量中精準獲得主控因子。在主影響因素評價中,僅選擇少量指標,對主要影響因子很難作出準確合理的抉擇。因此,在不同的研究中,應圍繞核心的研究目標,謹慎選取解釋變量,并盡可能多地獲得解釋變量個數,以獲得更為真實的研究結果。此外,在數據有限的情況下,如何快速高效地找出具代表性的環境變量因子也應是未來研究的重要內容之一。
4.2進一步改進冗余分析方法
隨著分析軟件的不斷發展,新的排序方法不斷出現,為能滿足相關生態學研究人員對于多元數據深入挖掘分析的需求。還需對該軟件系統作進一步改進。為減少研究人員因軟件模塊多而增加分析工作量負擔,需整合該軟件使其不再有分離的模塊,改進為集數據管理、分析和繪圖為一體的單一程序。數據輸入成功與否對進行下一步往往是初學者最大的障礙,需簡化數據輸入,直接導入數據即可進行分析是未來發展的趨勢所向。而在最新版本中已實現并提供一套完善簡單的繪圖工具,簡化方差分解和顯著性檢驗的操作步驟,增加單個解釋變量的顯著性檢驗功能,增加群落內類群功能性狀與譜系關聯分析等[39]。因此,對于大部分病原菌、病毒未知因素的整個生態環境而言,該分析軟件具有實際應用價值,仍需進一步改進發展。
4.3尋找病原菌、病毒主控因子
微生物的研究對于人類理解整個生物圈的能量流通和物質循環、了解微生物在整個生態系統中的功能、挖掘有用的潛在微生物資源以及保護生物安全方面有著非常重要的意義。目前,物種的功能性狀與譜系分析是生態學研究的熱點之一。冗余分析法分析功能多樣性從探究陸地、水生生態系統中包括植物、動物、菌類,并已過渡至生物安全有關的疾病研究等不同營養級的生物體在群落和生態系統中的功能及其范圍。但在微生物相關疾病研究中應用較少,病原菌、病毒與環境因素間的影響關系尚不清楚。尤其是研究暴露于環境中的病原微生物的功能性狀、核心群組、譜系地理分布與各因素的關系,有望為解決該分析對尋找微生物領域潛在致病菌的主控影響因子提供極具價值的線索,為流行疾病的預防提供參考。如,哪些影響因子可以抑制或促進病原菌或病毒的擴散和變異,這些結果可能對臨床試驗提供一定的參考意義。根據基因、遺傳、性狀與環境數據探索這些物種的致病因素將成為未來的發展趨勢。
4.4多分析方法的有機結合
要實現全面,準確的微生物與環境間影響關系的評價,任何一種單一的分析軟件都無法完成。盡管冗余分析方法提供了快速獲得解釋變量與響應變量間關系的解決方案,但該方法仍屬于單一分析方法,其核心算法模型較為單一,可能獲得的理論結果與實際情況具有一定的差異。因此,隨著生態學處理數據相關軟件功能的升級和完善,需綜合考慮使用多軟件(R語言、Python)、多分析方法(網絡分析、結構方程模型)與冗余分析的結果進行對比分析,進而獲得更為真實的結果。
參考文獻:
[1]Franklin J,Wiser S K,Drake D R,et al.Environment disturbance history and rain forest composition across the islands of Tonga,Western Polynesia[J].Journal of Vegetation Science,2006,17:233244.
[2]Sharma G P,Raizada P,Raghubanshi A S.CANOCO reference manual and CANOCO Draw for windows users guide: software for canonical community ordination (version 4.5)[J].Weed Biology and Management,2009,9(3):185191.
[3]Thibaut C,Keurcien L,Blum M G B,et al.Evaluation of redundancy analysis to identify signatures of local adaptation[J].Molecular Ecology Resources,2018,18(6):137.
[4]Legendre P,Legendre L.Canonical analysis[J].Developments in Environmental Modelling,2012,24: 625710.
[5]Terbraak C J F,Smilauer P.CANOCO reference manual and user's guide:software for ordination,version 5.0[J].New York:Microcomputer Power Ithaca,2012.
[6]Meng C,Liu H Y,Li Y Y,et al.Influences of the landscape pattern on riverine nitrogen exports derived from legacy sources in subtropical agricultural catchments[J].Biogeochemistry,2021,2:117.
[7]Liu Y,Zhang X,Yang M L,et al.Study on the correlation between soil microbial diversity and ambient environmental factors influencing the safflower distribution in Xinjiang[J].Journal of Basic Microbiology,2020,60(6):115.
[8]Yang F,Wu J J,Zhang D D,et al.Soil bacterial community composition and diversity in relation to edaphic properties and plant traits in grasslands of Southern China[J].Applied Soil Ecology,2018,128:4353.
[9]Cao B,Zhang Y,Wang Z Y,et al.Insight into the variation of bacterial structure in atrazinecontaminated soil regulating by potential phytoremediator:Pennisetum americanum (L.) K.Schum[J].Frontiers in Microbiology,2018,9:111.
[10]Wang X L,Yu S Q,Zhou L X,et al.Soil microbial characteristics and the influencing factors in subtropical forests[J].Acta Ecologica Sinica,2016,36(1):815.
[11]朱華德.五龍池小流域土壤水分時空變異及其與主要影響因子的關系[D].武漢:華中農業大學,2014:1125.
[12]任玉連,陸梅,曹乾斌,等.南滾河自然保護區森林土壤酶活性對海拔升高的響應[J].林業科學,2020,56(4):2234.
[13]任玉連,陸梅,范方喜,等.高原濕地沼澤化草甸土壤真菌與理化性質的關系[J].生態科學,2019,38(1):4249.
[14]周欣,左小安,趙學勇,等.科爾沁沙地植物群落分布與土壤特性關系的DCA、CCA及DCCA分析[J].生態學雜志,2015,34(4):947954.
[15] 王桂紅,何尋陽,蘇以榮,等.玉米和牧草典型種植模式下不同土層中土壤微生物量分布差異的研究[J].山地農業生物學報,2018,37(4):5157.
[16]秦杰,姜昕,周晶,等.長期不同施肥黑土細菌和古菌群落結構及主效影響因子分析[J].植物營養與肥料學報,2015,21(6):15901598.
[17]Shu D T,He Y L,Yue H,et al.Microbial structures and community functions of anaerobic sludge in six fullscale wastewater treatment plants as revealed by 454 highthroughput pyrosequencing[J].Bioresource Technology,2015,186:163172.
[18]Cui H,Ou Y,Wang L X,et al.Dynamic changes in microbial communities and nutrient stoichiometry associated with soil aggregate structure in restored wetlands[J].Catena,2021,197:104984.
[19]于小彥,張平究,張經緯,等.城市河流沉積物微生物量分布和群落結構特征[J].環境科學學報,2020,40(2):585596.
[20]裴振,孔強,郭篤發.鹽生植被演替對土壤微生物碳源代謝活性的影響[J].中國環境科學,2017,37(1):373380.
[21]王蕓,歐陽志云,鄭華,等.中國亞熱帶典型天然次生林土壤微生物碳源代謝功能影響因素[J].生態學報,2012,32(6):18391845.
[22]Fierer N,Jackson R B.The diversity and biogeography of soil bacterial communities[J].Proceedings of the National Academy of Sciences of the United States of America,2006,103(3):626631.
[23]Fierer N,Mccain C M,Meir P,et al.Microbes do not follow the elevational diversity patterns of plants and animals[J].Ecology,2011,92(4):797804.
[24]曹聰,阮超越,任寅榜,等.模擬增溫對武夷山不同海拔森林表層土壤碳氮及酶活性的影響[J].生態學報,2020,40(15):53475356.
[25]Lemanceau P,Blouin M,Muller D,et al.Let the core microbiota be functional[J].Trends in Plant Science,2017,22(7):583595.
[26]董醇波,張芝元,韓燕峰,等.核心微生物組的研究及利用現狀[J].菌物學報,2019,38(1):110.
[27]Mukhtar S,Mirza B S,Mehnaz S,et al.Impact of soil salinity on the microbial structure of halophyte rhizosphere microbiome[J].World Journal of Microbiology and Biotechnology,2018,34(9):117.
[28]Meng L,Wang J,Li X,et al.Microbial community and molecular ecological network in the EGSB reactor treating antibiotic wastewater:response to environmental factors[J].Ecotoxicology and Environmental Safety,2021,208:111669.
[29]魯少文,魏翠翠,李澤華,等.新型己酸菌強化對人工窖泥培養過程中原核微生物群落結構及酸、酯含量的影響[J].應用與環境生物學報,2020,26(1):144151.
[30]Yue Y,Shao T Y,Long X H,et al.Microbiome structure and function in rhizosphere of Jerusalem artichoke grown in saline land[J].Science of the Total Environment,2020,724:116.
[31]Bergstrom A K,Karlsson J,Karlsson D,et al.Contrasting plankton stoichiometry and nutrient regeneration in northern arctic and boreal lakes[J].Aquatic Sciences,2018,80(2):1424.
[32]Zhang H H,Zong R R,He H Y,et al.Biogeographic distribution patterns of algal community in different urban lakes in China: insights into the dynamics and coexistence[J].Journal of Environmental Sciences,2021,100:216227.
[33]Liu J,Ma D,Ma L,et al.Geographic distribution pattern of low and high nucleic acid content bacteria on a rivercatchment scale[J].Marine and Freshwater Research,2017,68:1055.
[34]劉文潔,王洋,周禮紅,等.貴州地區肥胖人群腸道微生物群落特征分析[J].山地農業生物學報,2020,39(1):919.
[35]Kamierczak S K,Ruszkowski J,Fic M,et al.Saccharomyces boulardii CNCM I745:a nonbacterial microorganism used as probiotic agent in supporting treatment of selected diseases[J].Current Microbiology,2020,77(9):110.
[36]Mazhar S H,Li X,Rashid A,et al.Coselection of antibiotic resistance genes,and mobile genetic elements in the presence of heavy metals in poultry farm environments[J].Science of The Total Environment,2021,755:142702.
[37]Guo H H,Xue S H,Nasir M,et al.Impacts of cadmium addition on the alteration of microbial community and transport of antibiotic resistance genes in oxytetracycline contaminated soil science direct[J].Journal of Environmental Sciences,2021,99:5158.
[38]Zhang H H,Wang Y,Chen S N,et al.Water bacterial and fungal community compositions associated with urban lakes,Xi’an,China[J].International Journal of Environmental Research and Public Health,2018,15(3):469.
[39]賴江山.生態學多元數據排序分析軟件Canoco 5介紹[J].生物多樣性,2013,21(6):765768.
猜你喜歡
商情(2016年42期)2016-12-23 14:25:52
商情(2016年42期)2016-12-23 13:35:35
東方教育(2016年4期)2016-12-14 22:15:13
財經界·學術版(2016年19期)2016-11-16 16:28:33
科技視界(2016年21期)2016-10-17 17:37:34
中國實用醫藥(2016年24期)2016-10-17 04:31:12
中國實用醫藥(2016年24期)2016-10-17 03:37:40
中國實用醫藥(2016年24期)2016-10-17 03:35:06
科學與財富(2016年28期)2016-10-14 21:58:50
