離子液體在放射性核素標記中的應用
2022-03-15 02:17:28褚泰偉
核化學與放射化學 2022年1期
關鍵詞:體系
王 帆,褚泰偉
北京大學 化學與分子工程學院,放射化學與輻射化學重點學科實驗室,北京 100871
通常所講的離子液體是一種由有機陽離子與無機或有機陰離子所組成的有機鹽,在室溫或室溫附近呈液態,所以又叫室溫離子液體[1-2]。離子液體具有良好的穩定性、可設計性,幾乎可以忽略的蒸汽壓、較寬的電化學窗口、不可燃、無毒等特點,被認為是新一代的綠色化學溶劑[1,3-5]。由于其特殊的物理化學性質,離子液體已經在合成催化、分離純化、電化學等諸多領域受到廣泛的關注[4,6-11]。近年來,關于離子液體在生命科學和醫藥等領域的研究逐漸增多,人們開始關注離子液體的生物活性以及其在醫藥等領域轉化應用的潛力[12-13]。一些關于離子液體應用的新領域逐漸拓展,例如,離子液體可以直接作為抗菌藥、抗癌藥,或在藥物遞送過程中作為佐劑成分等[14-17]。
放射性藥物是用于診斷、治療或醫學研究的放射性核素制劑或其標記藥物或生物制劑。與普通的藥物分子不同,由于放射性核素半衰期、穩定性等原因的限制,除一些簡單的化合物(如[131I] NaI口服溶液)外,放射性藥物分子一般很難以成品的形式儲存使用。大部分放射性藥物均是現標記現使用,尤其是診斷用放射性藥物。其中,標記前的化合物分子一般被稱為標記前體。標記前體通常由兩部分組成,即可以實現靶向功能的生物活性基團以及能夠結合放射性核素的標記位點。如何實現標記前體以較高的放射化學產率和放射化學純度的快速標記,對于推動放射性藥物在臨床上的使用至關重要。因此,發展快速、安全、高效的放射性標記方法是該領域學者一直追求的目標。按照放射性標記的核素種類來分,可以將放射性藥物劃分為非金屬類核素標記藥物和金屬類核素標記藥物。前者的標記主要通過涉及有共價鍵形成的有機反應來完成,特別是取代反應;而后者的標記則主要依賴于金屬離子與合適配體間的配位作用來完成。因此,有機化學和配位化學等學科領域的突破對于推進發展新的放射性標記方法學具有重要意義。多年來,離子液體已廣泛應用于有機化學以及配位化學領域,發展和積累了一系列新的研究工作。現有報道的離子液體體系中的放射性核素標記反應和所涉及到的核素種類比較有限,但這方面的研究無疑是一個具有較大研究潛力的新方向,也非常值得關注。本文主要介紹了幾種醫用放射性核素或與其相關的穩定同位素在離子液體體系中的標記方法,旨在拓寬放射性藥物化學中離子液體應用的新思路。
1 離子液體在放射性非金屬標記中的應用
常見的用于核醫學領域的非金屬核素主要有18F、131I、125I、11C等,關于這些核素在離子液體中的放射性標記報道較少,僅在18F、125I的標記中有少量提及,該領域的工作有待進一步深入研究。
1.1 離子液體在18F標記中的應用
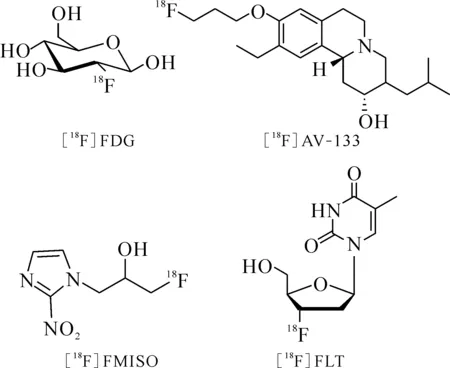
圖1 [18F]FDG、[18F]AV-133、[18F]FMISO和[18F]FLT的化學結構Fig.1 Structures of [18F]FDG, [18F]AV-133, [18F]FMISO and [18F]FLT
1.1.118F標記常規方法18F由于其合適的半衰期(T1/2=109.8 min),適宜的正電子能量,是目前用于核醫學顯像的最重要核素之一。大量重要的18F標記化合物被用于臨床診斷,如2-[18F]氟-2-脫氧-D-葡萄糖([18F]FDG)、(2R,3R,11bR)-9-(3-[18F]氟丙氧基)-3-異丁基-10-甲氧基-2,3,4,6,7,11b-六氫-1H-吡啶并[2,1-a]異喹啉-2-醇([18F]AV-133)、1-H-1-(3-[18F]氟-2-羥基丙基)-2-硝基咪唑([18F]FMISO)、3′-脫氧-3′-[18F]氟代胸腺嘧啶([18F]FLT)等,其結構式示于圖1。鑒于18F的半衰期相對較短,標記反應需要在較短時間完成并獲得可以滿足臨床轉化需求的放射化學產率和放射化學純度。目前已報道的用于合成18F標記化合物的標記反應有很多,按其反應機理來分主要有親電氟化反應、親核氟化反應等[18-22]。此外,用于18F標記的電化學氟化反應也有所報道[23]。親電氟化反應是最早研究的一類氟化反應,目前廣泛應用于臨床的糖代謝顯像劑[18F]FDG最早就是在一氟三氯甲烷(CFCl3)中通過與[18F]F2親電加成反應生產的[24-25],反應式示于圖2。[18F]F2是最簡單的親電氟化試劑,但以[18F]F2作為氟化試劑進行親電氟化反應只能利用一半的18F,放射性核素利用率低。在制備[18F]F2時往往需要引入大量穩定同位素[19F]F2氣體,導致最終制備的標記化合物比活度較低。由于F2具有很高的反應活性,反應的區域選擇性較差,放射化學產率較低,常伴隨著較多的副產物,產物較難純化。此外,F2還具有較強的腐蝕性,對于反應設備的要求較高,大多數的生產單位不具備這樣的硬件設備,使得這類反應在臨床上轉化非常困難。盡管目前一些較為溫和的親電氟化反應試劑也有所報道,但它們通常對反應底物要求較高,且較難獲得其相應的放射性氟源。相對于放射性親電氟化反應,以18F-作為氟源的親核氟化反應則是目前廣泛使用的18F標記策略。18F-可通過回旋加速器轟擊重水靶(H218O)產生[26],經過陰離子交換樹脂完成18F-的捕獲富集后便可用于標記反應。傳統的18F-親核標記通常選用干燥的非質子性溶劑作為反應溶劑[27],如無水乙腈、二甲基亞砜(DMSO)等。在標記反應開始前,從陰離子交換柱上洗脫下來的18F-會用無水乙腈進行共沸干燥2~3次,降低反應體系中水的含量,減弱18F-與水之間強的氫鍵作用,提高18F-的親核性。此外,18F-在有機溶液中的溶解度較低,為增加18F-在有機相中的溶解度,反應體系中常需要添加合適的相轉移催化劑以增加親核氟化反應的活性(如[18F]FDG的親核氟化合成途徑[28],反應式示于圖3)。相對于親電氟化反應來說,18F-的親核氟化反應對設備需求較低,氟源獲取相對簡單、容易,反應類型可以滿足大多數化合物的標記需求。這類反應更易于在臨床上得到轉化。但目前這類反應的反應條件一般較為劇烈,放射化學產率較低,操作繁瑣,放射性合成周期較長。尤其是共沸干燥步驟會花費大量的時間。同時,無水溶劑中18F的“裸離子”在反應中除了作為親核試劑以外,還具有一定的堿性,對于一些較為敏感的前體分子可能會有消去和羥基化等副產物的產生。從放射性18F的來源,實際應用過程中的安全性、操作性和設備需求等多角度出發考慮,相對于18F親電氟化標記策略,將18F親核氟化策略用于放射性藥物的合成更加具有現實意義。發展新的放射性18F親核氟化方法對于推動18F標記藥物在臨床上的轉化工作至關重要。
1.1.2離子液體體系中的親核氟化反應 基于離子液體的特殊性質,一些在離子液體中的新型親核氟化反應引起了關注[29]。盡管這些親核氟化反應并沒有全部在放射性18F標記中得到應用,但這些工作對于發展新的18F親核標記方法具有啟示意義。
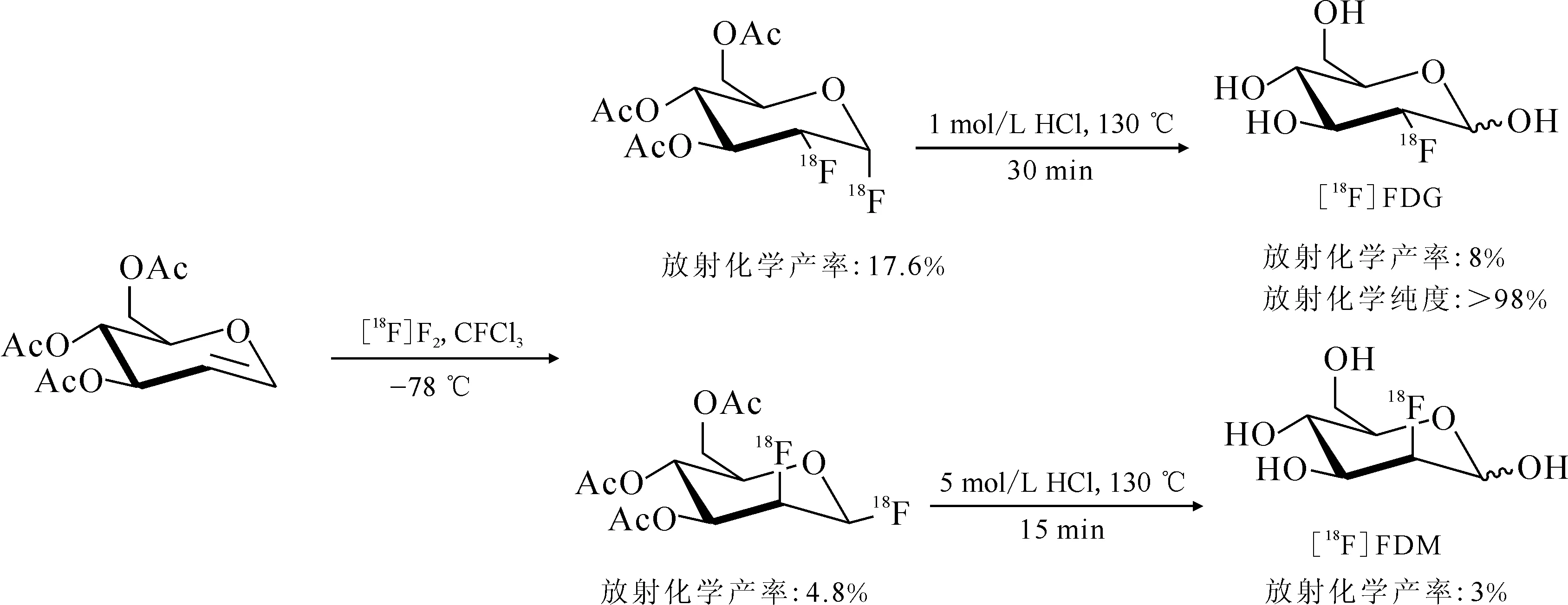
圖2 [18F]FDG親電氟化反應合成途徑[25]Fig.2 Synthesis of [18F]FDG by electrophilic fluorination[25]
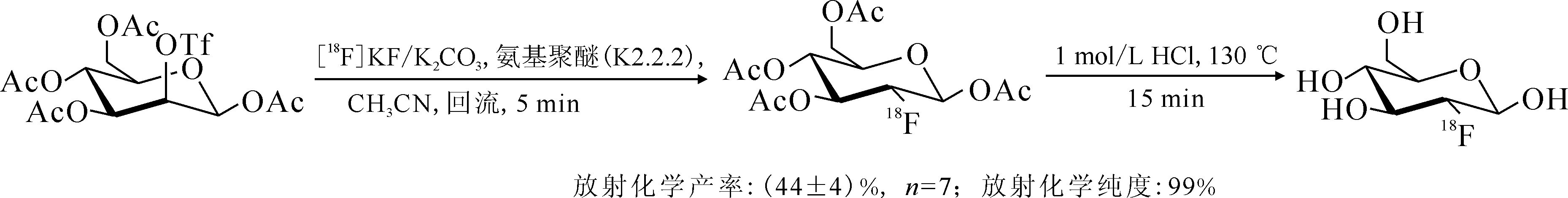
圖3 [18F]FDG親核氟化反應合成途徑[28]Fig.3 Synthesis of [18F]FDG by nucleophilic fluorination[28]
(1) 離子液體體系中氫鍵促進的親核氟化反應
一般認為,在質子溶液體系中,由于氫鍵的作用,F-的親核能力會受到很大的阻礙[30]。因此,氟化反應中的溶劑大部分時候會選用非質子溶劑,以保證F-的親核能力。基于這種原因,通常在18F親核標記前總是需要對體系進行干燥除水。然而,一些研究結果表明,溶液體系中存在的氫鍵不總是有礙氟化反應的進行[31]。
2002年,Kim等[32]首次報道了甲磺酸酯在離子液體和水存在下的親核氟化反應,反應式示于圖4。反應以KF為氟源,可在較為溫和的條件下將反應物轉化為相應的氟代化合物。有趣的是,此反應過程中少量水的存在對反應有促進作用。離子液體-水體系的使用不但提高了反應活性,更減少了副產物的形成。
作為該工作的補充,Kim等[33]進一步研究了甲磺酸酯在1-丁基-3-甲基咪唑四氟硼酸鹽([Bmim][BF4])體系中與多種金屬氟化物的氟化反應,反應式示于圖5。結果表明,CsF在堿金屬氟化物中的反應活性最強,而堿土金屬和過渡金屬氟化物在相同條件下則很難發生氟化反應。
隨后,Kim等[34]將該方法應用于18F標記化合物的合成,成功高效地實現了前體化合物的放射性標記,反應式示于圖6。反應在1-丁基-3-甲基咪唑三氟甲磺酸鹽([Bmim][OTf])和Cs2CO3存在下放射化學產率最高,且少量水的存在并不影響產物的高效合成。該方法的應用可省略傳統18F親核標記中耗時的共沸干燥過程,為后續的純化過程節約了大量的時間,更加具有臨床轉化價值。
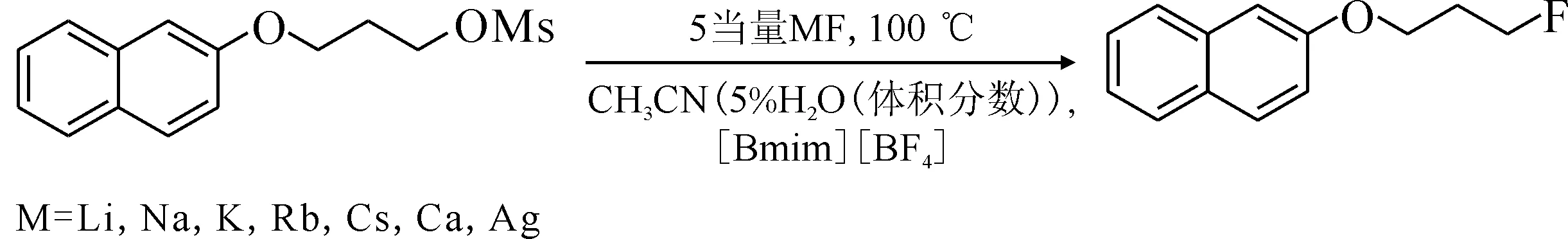
圖5 堿金屬氟化物、堿土金屬氟化物以及過渡金屬氟化物在離子液體和水存在下與甲磺酸酯的親核氟化反應[33]Fig.5 Fluorination of mesylate with alkali metal fluoride, alkaline earth metal fluoride and transition metal fluoride in presence of ionic liquid and water[33]

圖4 甲磺酸酯和KF在離子液體和水存在下的親核氟化反應[32]Fig.4 Nucleophilic fluorination of mesylate with KF in presence of ionic liquid and water[32]
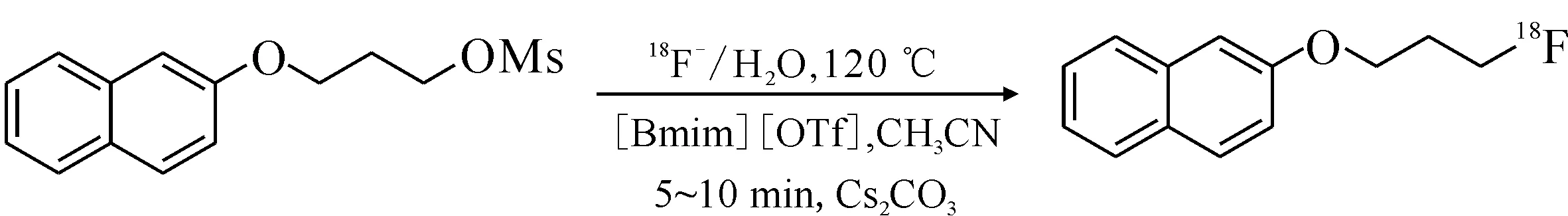
圖6 甲磺酸酯在[Bmim][OTf]中的18F標記反應[34]Fig.6 18F-labelling of mesylate in [Bmim][OTf][34]
在此基礎上,Kim等[35]成功將該方法應用于[18F]FDG的放射性快速合成,反應式示于圖7。在合成[18F]FDG的過程中,作者使用含[Bmim]-[OTf]的乙腈作為反應溶劑,省略了標記前的共沸干燥過程,縮短了[18F]FDG標記反應的時間,并獲得了較好的放射化學產率。
基于相同的方法,Moon等[36]報道了在[Bmim][OTf]中[18F]FLT的放射性合成,反應式示于圖8。該合成過程省略了乙腈的共沸干燥步驟,減少了前體分子的用量,縮短了標記反應的時間并減少了副產物的產生。從放射性合成開始到高效液相色譜(HPLC)純化結束,總時長約為70 min,產物的放射化學產率和放射化學純度良好,該合成方法下的放射化學產率可滿足日常自動化合成的需要。
(2) 離子液體體系與叔醇在親核氟化反應中的協同效應
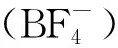
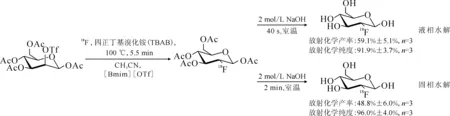
圖7 [18F]FDG在[Bmim][OTf]中的快速合成[35]Fig.7 Rapid synthesis of [18F]FDG in [Bmim][OTf] [35]
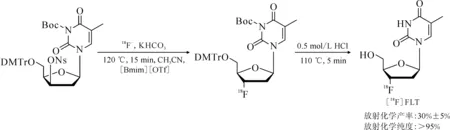
圖8 [18F]FLT在[Bmim][OTf]中的快速合成[36]Fig.8 Rapid synthesis of [18F]FLT in [Bmim][OTf][36]
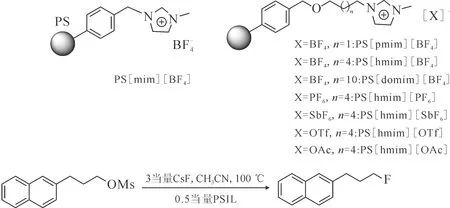
圖9 PSIL的結構式及甲磺酸酯在PSIL催化下的氟化反應[37]Fig.9 Structures of PSIL and fluorination of mesylate catalyzed by PSIL[37]
作為此工作的延續,Kim等[38]還報道了PSIL與叔醇在親核氟化反應之中的協同作用,反應式示于圖10。反應以堿金屬氟化物作為氟源,可以高效地將磺酸酯和鹵代烴類化合物轉化為相應的氟代產物。這主要是因為體系在保持氟離子親核性的同時,溶液中氟與叔醇羥基氫之間弱的氫鍵作用降低了氟離子的堿性,從而減少了副產物的產生。PS[hmim]BF4/叔醇體系的使用提高了氟離子的親核活性并抑制了副反應的發生。
基于以上結果,Shinde等[39]報道合成了一種叔醇功能化的離子液體并將其用于親核氟化反應的催化劑,離子液體結構式與反應式示于圖11。實驗結果表明,這種功能化離子液體不僅增加了F-的親核反應活性,還顯著抑制了副反應的發生。該研究證明了叔醇功能化的離子液體對于催化親核氟化反應具有協同作用。
隨后,Shinde等[40]進一步深入研究了該類功能化離子液體對于其它親核取代反應的催化活性,擴大了該類離子液體的應用范圍,反應式示于圖12。其中,在[mim-tOH][OMs]體系中,即使在室溫條件下,鹵化、疊氮化、乙酰氧基化和氰化等常見的親核取代反應均有著較高的反應活性。這一類功能化離子液體的協同催化效應在制備18F標記化合物的應用中可能有著潛在應用價值。在此基礎上,Oh等[41]深入研究了離子液體中SN2親核氟化反應,提出了離子液體中SN2反應的機理,并通過理論計算解釋了[mim-tOH][OMs]功能化離子液體中協同效應的原因。

圖10 甲磺酸酯在PS[hmim]BF4/叔醇體系下的氟化反應[38]Fig.10 Fluorination of mesylate in PS[hmim]BF4/tertiary alcohol[38]
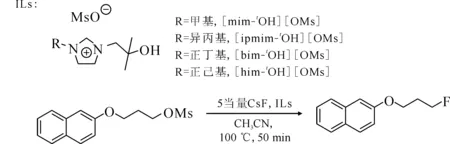
圖11 叔醇功能化離子液體的結構及甲磺酸酯在叔醇功能化離子液體中的氟化反應[39]Fig.11 Structures of tertiary alcohol functionalized ionic liquids and fluorination of mesylate in tertiary alcohol functionalized ionic liquids[39]

圖12 叔醇功能化離子液體中的親核取代反應[40]Fig.12 Nucleophilic substitution reactions in tertiary alcohol functionalized ionic liquids[40]
Shinde等[42]還將這種功能化的離子液體負載到了聚合物上,并研究了其在親核氟化反應中的催化活性,叔醇功能化離子液體負載聚合物結構式及催化氟化反應式示于圖13。實驗結果表明,與未功能化離子液體負載的聚合物相比,含叔醇功能化離子液體負載的聚合物表現出更加優異的催化活性和選擇性,這主要是由于咪唑鹽與叔醇之間的協同作用的結果。由于這種叔醇功能化離子液體中協同效應的存在,它們對于親核取代反應有著高的催化反應活性。
(3) 其它離子液體體系中的親核氟化反應
Murray等[43]報道了1-丁基-3-甲基咪唑六氟磷酸鹽([Bmim][PF6] )中,以CsF作為氟源的鹵素交換反應,反應式示于圖14。結果表明,反應性較強的底物如苯甲酰氯或芐溴的氟化反應可以在室溫下進行,而對于反應活性較低的溴代烷烴,會因消除反應作為競爭反應的存在而降低氟化產物的產率。
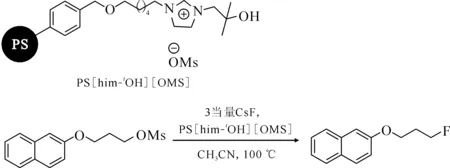
圖13 叔醇功能化離子液體負載的聚合物結構及甲磺酸酯在該體系下的親核氟化反應[42]Fig.13 Structures of polymer-supported tertiary alcohol functionalized ionic liquid and nucleophilic fluorination of mesylate in system[42]
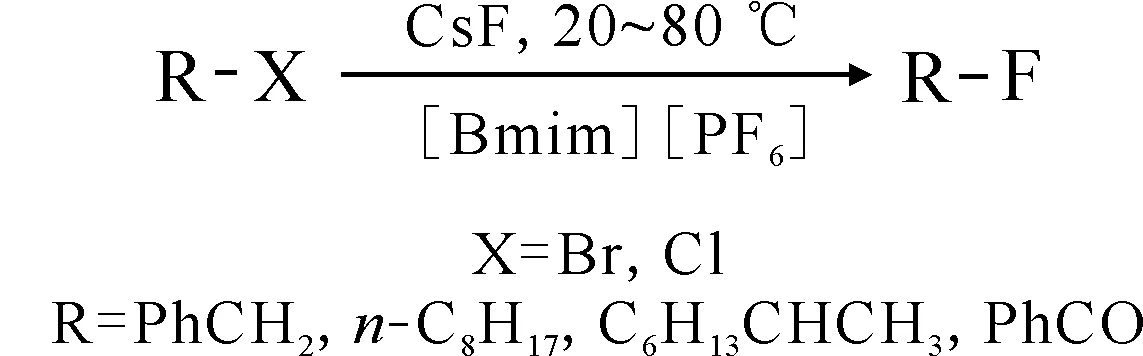
圖14 鹵代烴在[Bmim][PF6]中的親核氟化反應[43]Fig.14 Nucleophilic fluorination of halogenated hydrocarbons in [Bmim][PF6][43]
Zhong等[44]報道了第一例芳雜環化合物在離子液體中的親核氟代反應,反應式示于圖15。反應在[Bmim][BF4]中發生,以2,3,5-三氯吡啶為原料、堿金屬氟化物為氟源制備了5-氯-2,3-二氟吡啶。
Anguille等[45]報道了在離子液體體系中,KF與三氯甲苯的氟化反應,反應式示于圖16。研究表明,該反應比一般在有機溶劑中進行的反應速率更快,反應速率與離子液體中的陰離子有關。此外,作者還發現,離子液體體系中存在少量溴鹽,會強烈激活[Bmim][PF6]體系中的親核氟化。
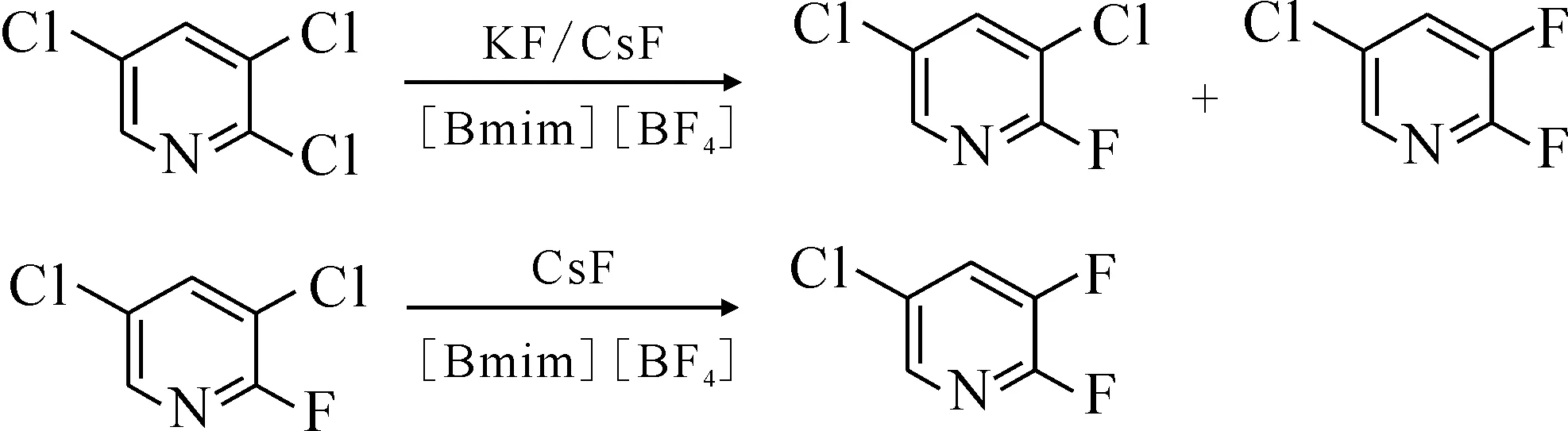
圖15 2,3,5-三氯吡啶在[Bmim][BF4]中的親核氟化反應[44]Fig.15 Nucleophilic fluorination of 2,3,5-trichloropyridine in [Bmim][BF4][44]

圖16 三氯甲苯在[Bmim][PF6]中的親核氟化反應[45]Fig.16 Nucleophilic fluorination of trichlorotoluene in [Bmim][PF6][45]
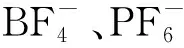
Chu等[47]報道了在[Bmim][BF4]體系下,通過Wallach反應可以將氟引入帶有吸電子取代基的芳烴之中,反應式示于圖18。在這類反應之中,離子液體既充當反應溶劑,又是氟化反應中的氟源。反應結束后,離子液體可通過HF處理再重復使用。
Paramanik等[48]設計合成了一種新的三烷基氧化膦功能化的咪唑離子液體,并將其應用到以CsF為氟源的甲磺酸伯烷基酯的親核氟化反應中,離子液體結構式及氟化反應式示于圖19。結果表明,與單獨的[Bmim][OMs]或三烷基氧化膦相比,該離子液體可以加速親核氟化反應的發生。氟化反應速率的提升可能是由于咪唑鹽和氧化膦官能團對氟化反應過程中的累積電荷綜合作用的結果,從而加速了反應。該反應同樣可以以鹵代烴作為反應底物,但以鹵代烴為底物會伴隨消去反應的發生,碘代烷尤為明顯。當底物具有對堿敏感的差向異構中心時,反應過程中應避免使用過量的氟鹽以防止底物手性的改變。該方法有望應用于以甲磺酸酯為前體的18F藥物的制備之中。
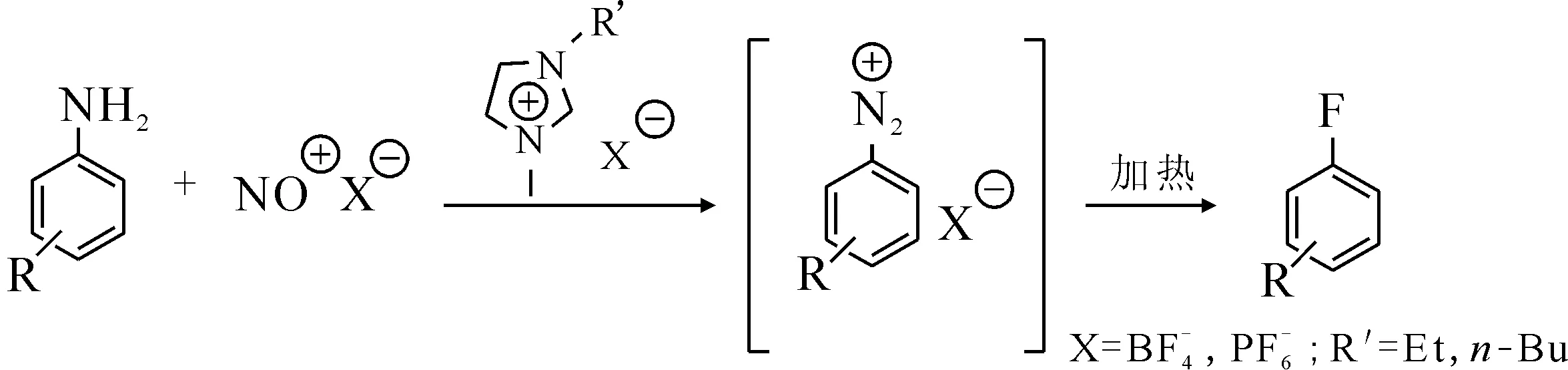
圖17 芳基重氮鹽在離子液體中的原位氟化反應[46]Fig.17 In-situ fluorination of aryl diazonium salt in ionic liquids[46]
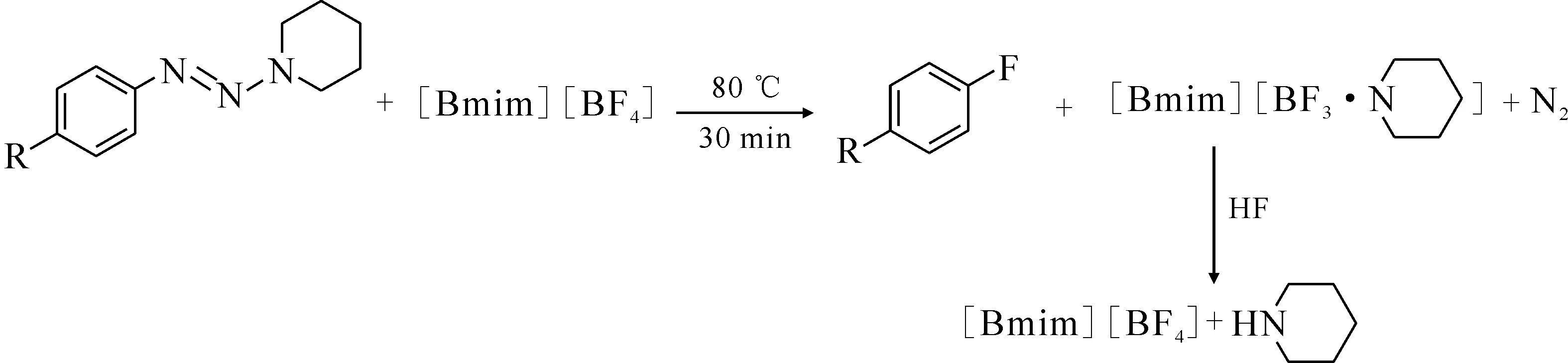
圖18 在[Bmim][BF4]中通過三氮烯的氟化合成氟代芳烴[47]Fig.18 Synthesis of fluoroaromatic hydrocarbons by fluorination of triazene in [Bmim][BF4][47]
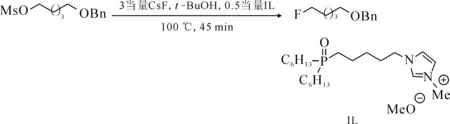
圖19 三烷基氧化膦功能化離子液體的結構及甲磺酸酯在該離子液體中的氟化反應[48]Fig.19 Structure of trialkylphosphine oxide functionalized ionic liquid and fluorination of mesylate in ionic liquid[48]
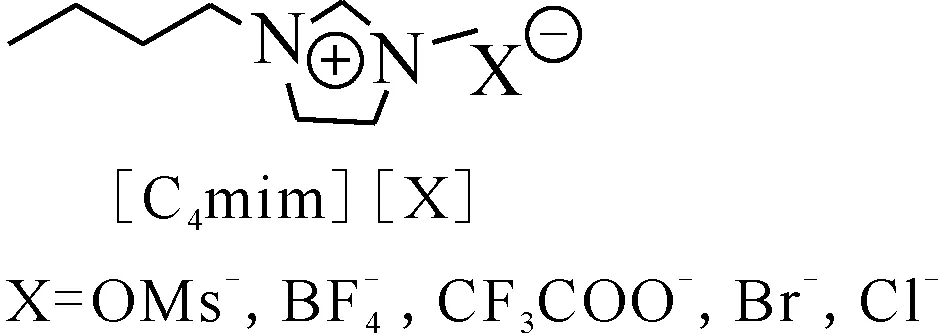
圖20 離子液體結構[49]Fig.20 Structures of ionic liquids[49]
相比于傳統有機溶劑體系,離子液體作為氟化反應的溶劑具有很大的優勢。得益于有機氟化學的發展,在離子液體體系中的氟化研究工作已有一些進展。盡管目前該體系下的親核氟化反應在放射性18F標記化合物合成中的應用還十分有限,但其無疑為發展新的18F親核標記方法提供了思路。
1.2 離子液體在碘化反應中的應用
放射性碘是臨床上使用較多的示蹤核素之一,它有著多種同位素,常見的核素有123I、124I、125I和131I等。關于離子液體體系中放射性碘或穩定碘的碘代反應的報道相對較少。
Racys等[50]報道了在離子液體體系中通過Fe(Ⅲ)催化活化N-碘代琥珀酰亞胺(NIS)實現了在溫和條件下芳烴快速碘化,反應式示于圖21。該碘化反應可在較低的反應溫度下進行,無需使用昂貴的貴金屬催化劑,具有較高的反應速率和區域選擇性,而作為反應溶劑和Fe(Ⅲ)活化試劑的離子液體易于回收使用。該碘化反應有望在重要的藥物分子合成中有所應用。
由于碳-金屬鍵的高反應活性,有機金屬衍生>物可以在溫和的條件下實現各種底物的快速轉化。這類反應具有較好的區域選擇性,對底物中存在的敏感官能團,如酯基等具有良好的耐受性。但是這類反應所使用的金屬通常為一些重金屬元素,反應結束后的重金屬殘留等缺點限制了這類試劑使用。Rajerison等[51]報道了一種離子液體修飾的有機錫試劑用于放射性鹵素的標記,反應式示于圖22。該標記反應條件溫和,反應迅速。在標記反應結束后,無需使用HPLC純化,通過簡單的分離純化步驟便可獲得高放射化學產率的放射性At或放射性碘標記的化合物。該標記方法降低了錫的殘留量,并節約了分離純化所需的時間,更有利于在臨床上轉化與應用。
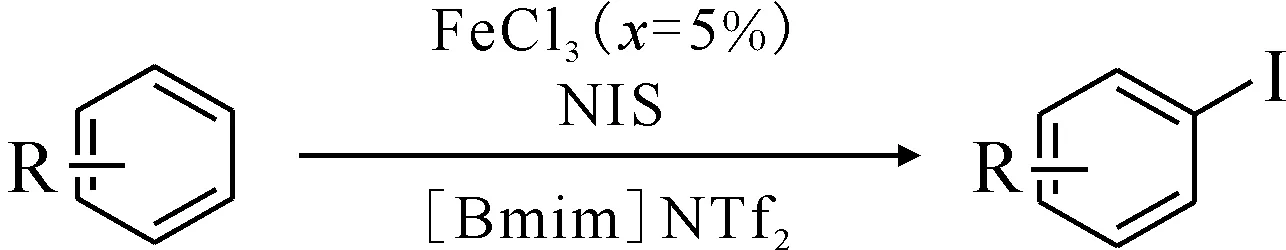
圖21 芳烴在[Bmim]NTf2中的碘化反應[50]Fig.21 Iodination of aromatic hydrocarbons in [Bmim]NTf2[50]

圖22 離子液體修飾的有機錫化合物用于放射性鹵素的標記[51]Fig.22 Organotin compounds modified by ionic liquids used for labelling of radioactive halogens[51]
2 離子液體在放射性金屬核素標記中的應用
目前關于離子液體在放射性金屬核素標記中的應用報道不多,僅僅在99Tcm和68Ga的放射性標記中有少量文獻報道。
2.1 離子液體在68Ga標記中的應用
68Ga具有優秀的核性質,半衰期為68 min,是用于正電子發射型計算機斷層顯像(PET)的重要核素,它被廣泛應用于蛋白和多肽化合物的放射性標記。68Ga的標記前體分子常以具有多個配位原子的螯合配體進行修飾,常見的配體如1,4,7,10-四氮雜環十二烷-1,4,7,10-四羧酸(DOTA)、1,4,7-三氮雜環壬烷-1,4,7-三乙酸(NOTA)等。一般來說,68Ga的標記反應通常需要在較高的反應溫度(70~100 ℃)以及合適的pH緩沖范圍下進行。在醋酸鹽或4-羥乙基哌嗪乙磺酸鹽(HEPES)的緩沖體系中的68Ga的放射性標記已經被廣泛報道,但歐洲藥典卻將HEPES定義為有毒雜質。最近,有學者合成了一些具有自我緩沖能力的離子液體,并將其作為一種緩沖溶液應用在68Ga標記反應之中。
Antuganov等[52]報道了一系列三乙醇胺(TEA)、三(羥甲基)甲基銨(TRIS)與具有生物活性的羧酸所形成的質子性離子液體,并將其作為一種緩沖溶液應用于68Ga-PSMA-HBED-CC(PSMA-11)標記反應中,離子液體結構式示于圖23。實驗結果表明,與常用的HEPES 緩沖液相比,在TEA和 TRIS緩沖體系下,標記反應可以使用相對較少的前體(5 μg),具備進一步研究的價值。TEA和 TRIS的緩沖體系可能是有毒的HEPES緩沖液的潛在替代品。
在此基礎上,Antuganov等[53]進一步研究發展了一系列N-苯基-二(2-羥乙基)胺(PDA)的羧酸鹽離子液體緩沖體系,離子液體結構式示于圖24。接著,作者系統性地研究了TEA、TRIS和PDA類離子液體在68Ga 放射性標記中的潛力。結果表明,在DOTA-NOC、DOTA-TATE、PSMA-617 以及 PSMA-11的68Ga 放射性標記中,使用合適的離子液體作為緩沖體系可以在低溫(t=37 ℃)下完成放射性標記。其中,三乙醇胺的苯甲酸鹽([TEA] Benz )和三乙醇胺的2-甲基苯氧乙酸鹽([TEA] Crez)是這一類緩沖溶劑中最好的,標記效率高于HEPES緩沖體系在相同實驗條件下的標記結果。同時,在該體系中添加有機溶劑對低溫放射性標記有著較大的影響,其中,丙酮是較好的共溶劑。
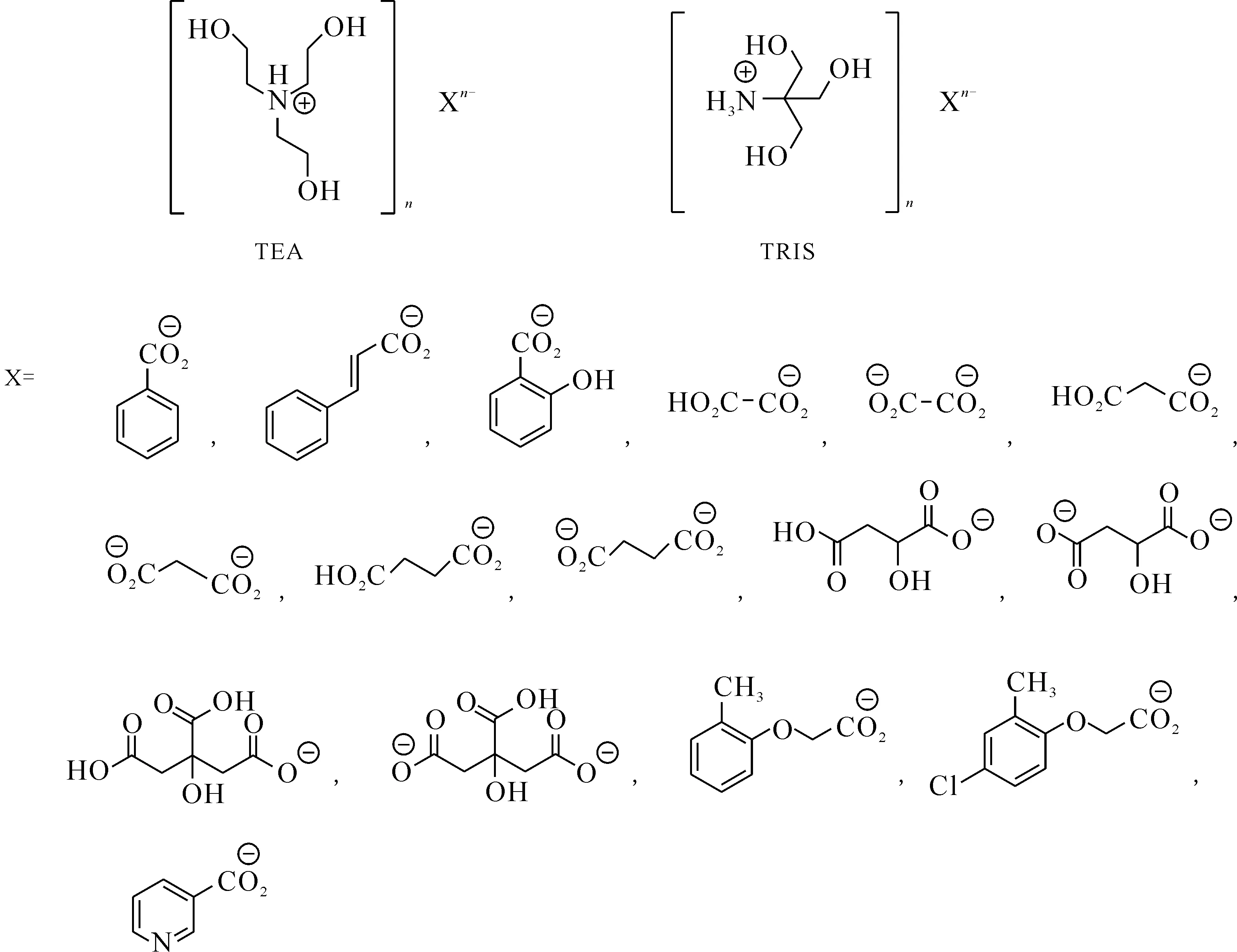
圖23 TEA、TRIS類離子液體的結構式[52]Fig.23 Structures of TEA and TRIS ionic liquids[52]
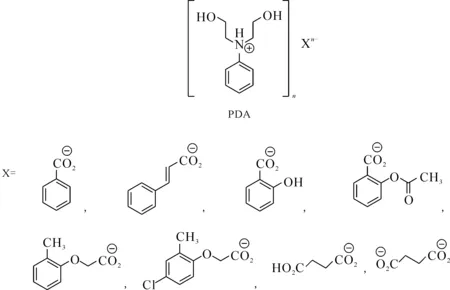
圖24 PDA類離子液體的結構式[53]Fig.24 Structures of PDA ionic liquids[53]
作為該工作的延續,Kondratenko等[54]進一步合成了一系列二乙醇胺(DEA)的羧酸鹽離子液體,離子液體結構式示于圖25,并對其在68Ga放射性標記過程中的緩沖作用進行了評估。結果表明,在苯甲酸二乙醇胺離子液體體系中,p-SCN-Bn-DOTA可以在高溫與低溫條件下均以高的放射化學產率與68Ga形成標記物。與HEPES作為緩沖體系的68Ga標記效率相比,苯甲酸二乙醇胺與多種標記前體的標記效率高于在相同條件下的HEPES緩沖體系下的標記效率。因此,苯甲酸二乙醇胺是一種極具潛力的放射性68Ga標記反應緩沖溶液,有著較大的臨床轉化價值。
圖25 DEA類離子液體的結構式[54]Fig.25 Structures of DEA ionic liquids[54]
2.2 離子液體在99Tcm標記中的應用
99Tcm是廣泛應用于核醫學顯像研究的放射性核素,價格低廉,可由99Mo-99Tcm發生器淋洗得到。在眾多的99Tcm標記的配合物分子中,99Tcm的雙芳烴配合物([99Tcm(arene)2]+)中有著容易修飾的苯環結構,具有發展成為新一代放射性藥物的潛力。
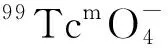
3 總結與展望
盡管目前離子液體在放射性藥物化學中的研究還不是很多,但現有的研究成果無不展示出其在放射性藥物化學中的應用潛力與價值。相信隨著離子液體在有機化學和配位化學等相關學科領域應用研究的繼續推進,會有越來越多的學者注意到離子液體在放射性藥物化學中的潛在應用,為放射性藥物化學學科的發展注入新的動力。
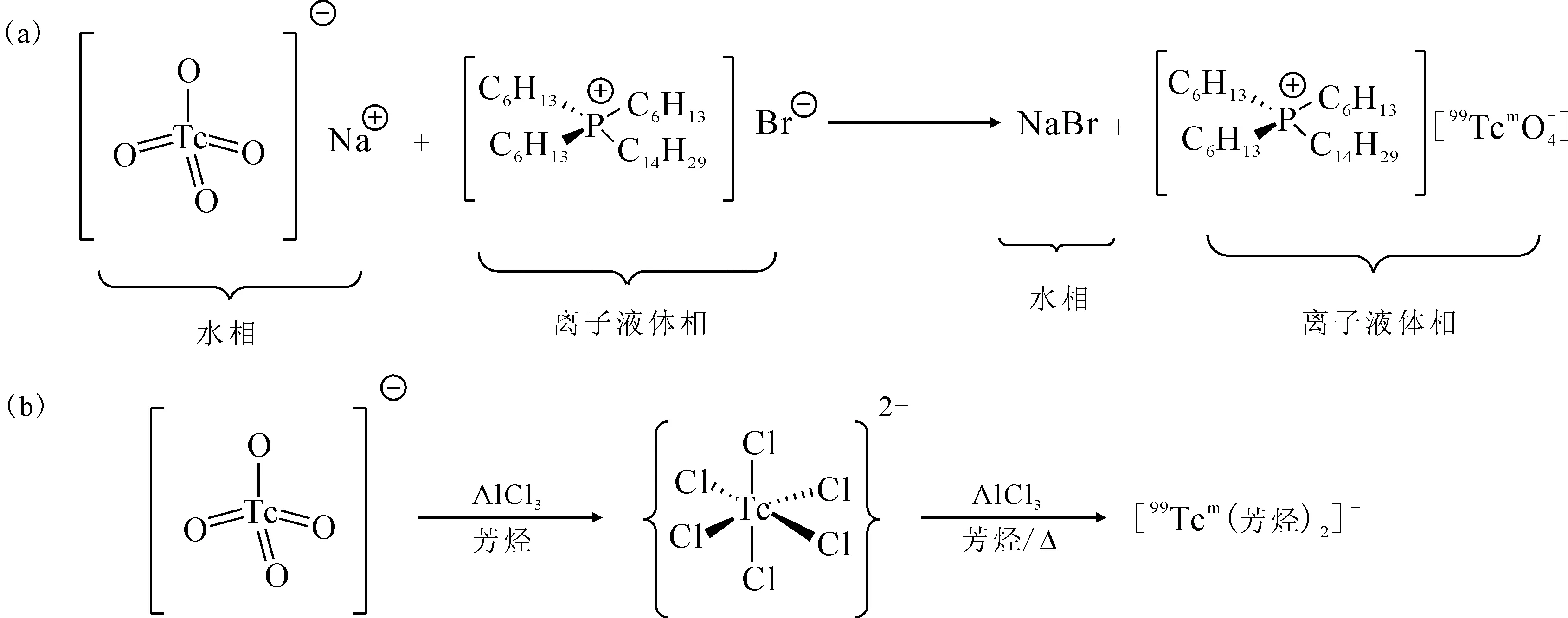
圖26 離子液體萃取機理(a)及雙芳烴锝配合物合成反應方程式(b)Fig.26 Mechanism(a) of extraction of by ionic liquid and synthesis(b) of bis-arene technetium complexes
猜你喜歡
商品與質量(2021年43期)2022-01-18 05:31:22
杭州(2020年23期)2021-01-11 00:54:42
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
中國外匯(2019年17期)2019-11-16 09:31:14
中國衛生(2015年12期)2015-11-10 05:13:40
現代企業(2015年1期)2015-02-28 18:43:18
汽車零部件(2014年5期)2014-11-11 12:24:28
新高考·高一物理(2014年1期)2014-09-18 01:26:07
浙江人大(2014年1期)2014-03-20 16:19:53
終身教育研究(2012年4期)2012-03-25 10:41:11
