腦血管病基因組學數據分析流程建設
2022-04-06 12:36:36許喆程絲劉陽石延楓李昊
中國卒中雜志 2022年3期
許喆,程絲,劉陽,石延楓,李昊
腦血管病是我國第一大死亡原因[1],且具有病因分型多[2]、遺傳架構(genetic architecture)復雜等特點[3]。隨著高通量測序和質譜技術的進步,在腦血管病分子機制研究中,基因組、轉錄組、表觀遺傳組、蛋白質組、代謝組、宏基因組等組學技術被應用[4-6],融合各組學技術的多組學分析已經成為未來醫學和生命科學研究的趨勢[7-8]。在眾多組學提供的不同維度的數據中,基因組信息在體內最為穩定,組織特異性低,因此成為多組學研究重要的切入點[7],應用也最為廣泛[9-15]。但是由于研究目標、樣本選擇有所不同,腦血管病領域各個基因組學研究的分析方案不盡相同,導致腦血管病領域基因組數據生物信息學分析流程整合度不高,缺乏系統、全面的匯總。
本研究在臨床需求和文獻調研的基礎上,梳理了腦血管病基因組學研究中常用、主流、穩定的分析方案,以模塊化的設計思路,以中國國家卒中登記Ⅲ(China national stroke registry-Ⅲ,CNSR-Ⅲ)研究的基因組學數據為測試集[16-17],搭建標準化的生物信息學分析流程,為腦血管病臨床和基礎研究提供大數據支撐。
1 方法
1.1 確定分析流程所需模塊和技術參數
1.1.1 文獻復習 通過查閱MEGASTROK E(multi-ancestry genome-wide association study of stroke)項目、GENS(GENetics of Stroke)登記研究、CHARGE(cohorts for heart and aging research in genomic epidemiology)合作組研究的相關文獻[15,18-19],總結腦血管病隊列和群體遺傳學研究中常用的分析策略、研究方案設計原則、數據質控方案和管理模式等,收集生物信息學分析的技術參數。
1.1.2 專家咨詢 咨詢神經病學、神經生物學、流行病學專家,調研臨床診斷和基因檢測對基因組學、生物信息學數據分析的需求。向生物信息學、群體遺傳學、基因組學、生物樣本庫等領域專家,咨詢基因組學數據分析中合理的功能模塊和實現方案。
1.1.3 數據庫和在線工具調研 調研生物信息學、基因組學、表觀遺傳學、轉錄組學等多組學數據庫,下載相應數據或者優化檢索方式,將數據庫的內容或信息嵌入分析流程。調研數據庫包括RegulomeDB[20]、GTEx(genotypetissue expression)[21]、GeneHancer(genomewide integration of enhancers and target genes in GeneCards)[22]、ClinVar(public archive of interpretations of clinically relevant variants)[23]、OMIM(online mendelian inheritance in man,https://omim.org)、Orphanet(www.orpha.net)等。同時調研了Galaxy等在線分析工具[24],考察基因組學數據標準化分析流程設計思路。
1.2 高性能運算集群 基于首都醫科大學附屬北京天壇醫院高性能運算集群,開發和搭建生物信息學分析流程。集群包括80臺1路、76臺2路、4臺4路、1臺8路CPU計算節點,9臺GPU計算節點,理論浮點運算能力375萬億次/秒。采用中科曙光parastor 200并行存儲系統,提供5.3 PB可用存儲空間。集群使用1套100 Gbps Infiniband網絡,1套千兆管理網絡,提供1臺登錄節點,提供用戶登錄、編譯軟件、提交作業、上傳下載數據等功能;提供1臺管理節點,安裝Gridview作業管理系統,并提供集群監控功能。
1.3 流程測試和優化 基于CNSR-Ⅲ研究等隊列的基因組學數據,對流程進行測試。測試樣本為CNSR-Ⅲ研究隊列遺傳亞組共12 603例缺血性腦血管病患者外周血白細胞DNA[17]。這些DNA樣本被用于進行全基因組測序,生成的遺傳數據經過本研究搭建的生物信息學流程處理,保留測序數據質量合格、無污染、無親緣關系、無顯著遺傳背景差異的樣本,用于后續的群體遺傳學、多組學數據挖掘。在測試過程中,基于高性能運算集群的架構和各個計算節點的算力,結合數據量、單個運算任務對內存需求、不同模塊中生物信息軟件的算法和并行計算的參數、集群帶寬等,拆分計算任務,使集群算力得到高效應用。
1.4 數據管理方案調研 遵照《中華人民共和國生物安全法》《中華人民共和國人類遺傳資源管理條例》及《首都醫科大學附屬北京天壇醫院人類遺傳資源管理辦法》的相關規定,對研究涉及的數據和樣本進行管理。與國內相關研究團隊的數據管理部門、相關專家進行學術交流,學習人類遺傳資源管理經驗。
2 結果
本研究搭建的基因組學數據生物信息學分析流程,主要包括組學大數據質控和預處理、群體遺傳學質控和樣本清理、臨床相關位點解析等部分,各部分又按照研究目的和功能、使用軟件和軟件組合的不同,細分為不同模塊(圖1)。不同模塊之間可以自由組合,增強了本流程的兼容性和普適性。此外,本流程在搭建過程中也考慮到研究方案頂層設計和項目管理的需求,在項目論證和規劃階段引入基因組學、生物信息學專家,以便明確研究目標,選擇合理的組學檢測技術。
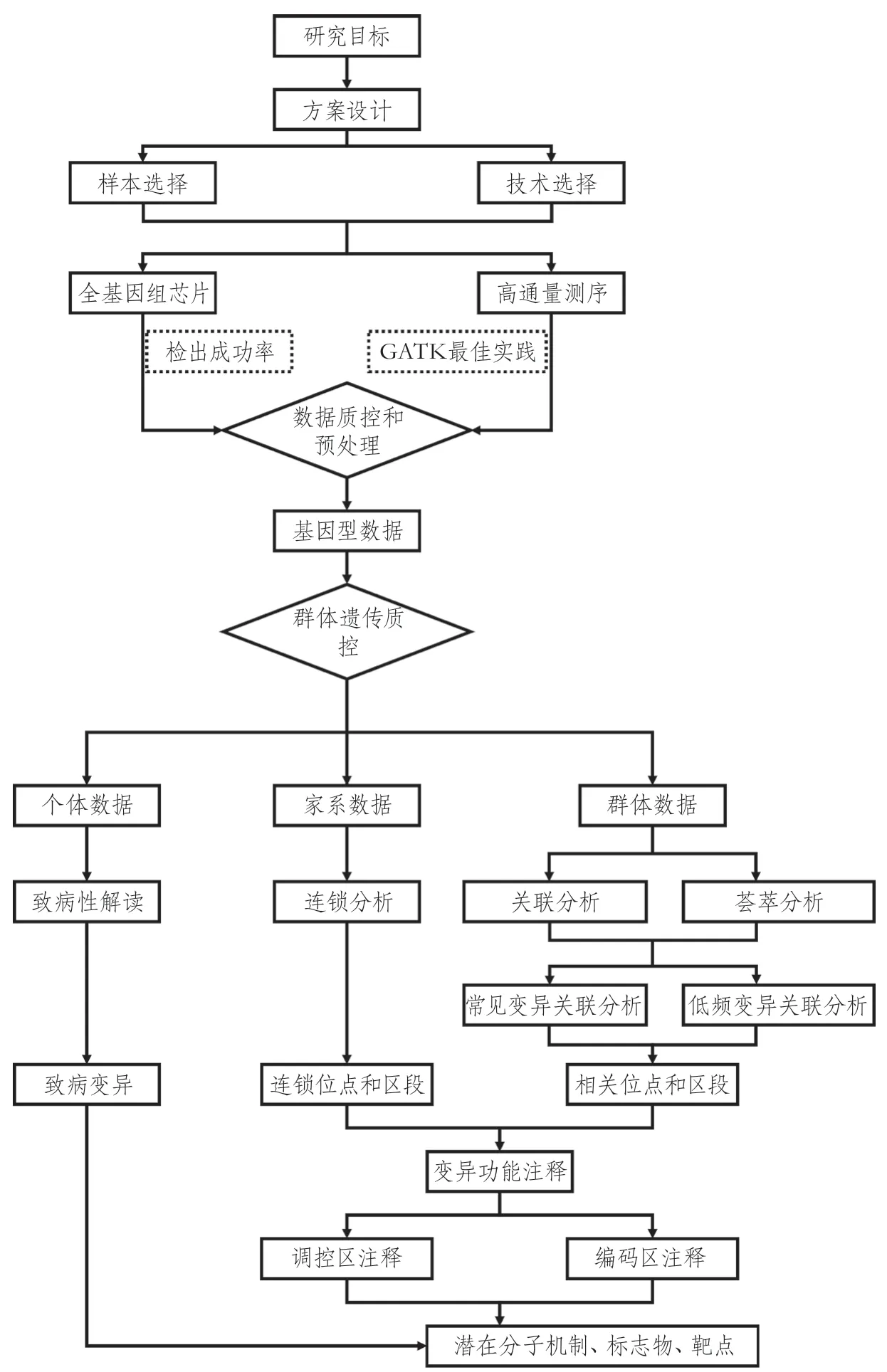
圖1 基因組學數據分析流程
2.1 檢測技術選擇 生物信息分析人員根據醫師科學家的研究目的,協商選取合適的檢測技術,預先確認分析方案。檢測技術的選擇遵循以下原則:①優先選擇數據質量可靠,穩定性、重復性好,認可程度高的檢測技術;②選擇樣本消耗量適中的檢測技術,兼顧數據和樣本用于后續其他研究的可能;③對于個體化訂制的檢測方案(如探針和靶向捕獲試劑盒等),需要進行預試驗,評估檢測體系運轉情況和數據質量,利用預試驗產生的基因組學數據,對分析流程進行測試和優化;④項目的檢測方法一旦確定,不宜中途調整,檢測試劑和儀器也應確保是同一型號、相同批次,避免檢測結果出現批次效應。
2.2 原始數據預處理與質量評估
2.2.1 高通量測序數據 高通量測序作為目前獲取樣本最全面遺傳信息的主流技術手段,被廣泛應用于復雜疾病的基因組學分析、孟德爾遺傳病的分子診斷等領域。通常情況下,高通量測序的實驗由專業的檢測機構或實驗室完成,檢測結果以測序原始數據(FASTQ格式)和檢測報告的形式向臨床研究人員反饋。
在測序原始數據復制、傳輸、備份的全部環節,都需要核對數據完整性,避免數據文件損壞。數據每復制一次,檢測原文件和復制本的MD5碼,兩文件的MD5碼必須完全一致。
而后,按照GATK(genome analysis toolkit)最佳實踐的高通量測序數據檢出種系變異中的單核苷酸變異(single nucleotide variants,SNV)和插入缺失多態(insertion-deletion,INDEL)的處理流程(圖2)[25],對測序原始數據進行預處理,同時完成測序深度、覆蓋情況、微生物污染(GC含量)、人源污染等方面的評估。
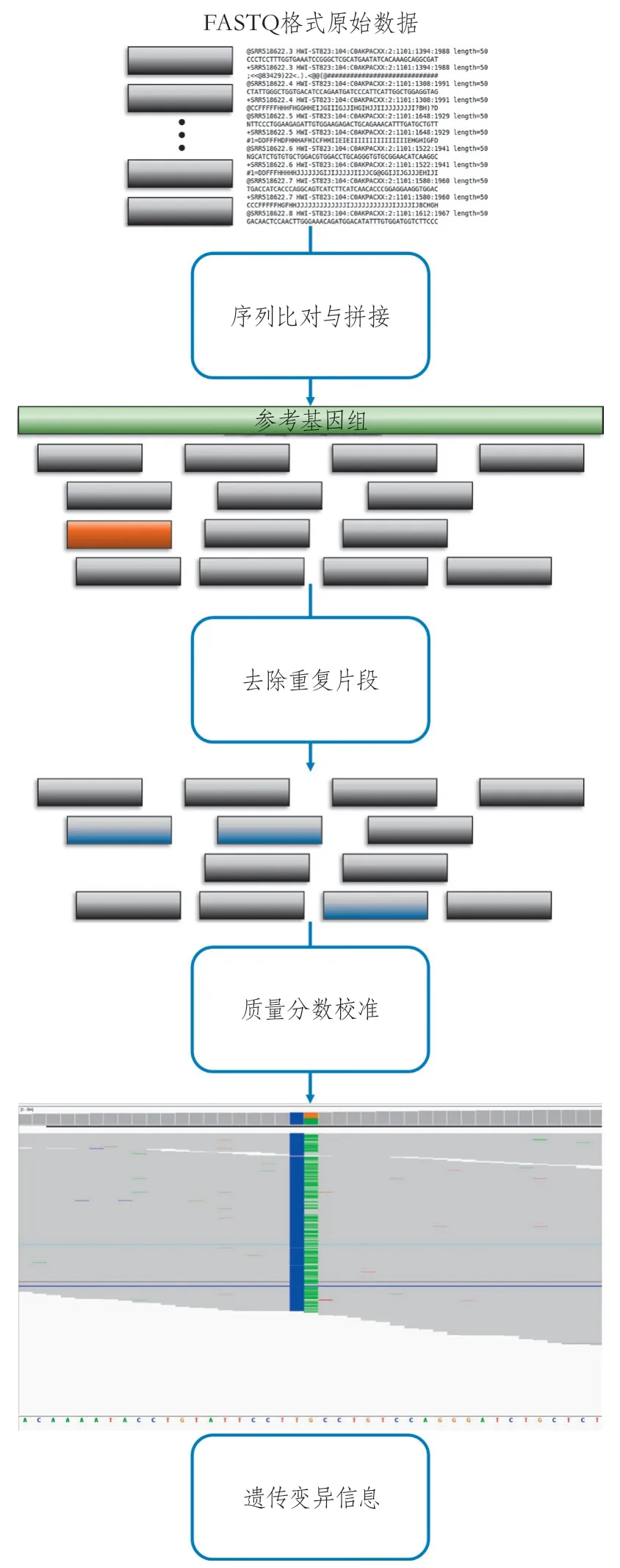
圖2 GATK最佳實踐流程
2.2.2 分型數據 分型是指獲取樣本預設變異位點基因型的操作,通常是指獲取單核苷酸多態(single nucleotide polymorphism,SNP)位點的基因型。根據通量和分型原理的不同,采用不同的方案進行預處理。
對于通量較高的全基因組SNP分型芯片,使用芯片廠家指定的軟件,如GenomeStudio(Illumina)和Genotyping Console(Affymetrix)進行SNP分型芯片的數據預處理。在生產廠商的網站找到相應型號芯片的解析文件,用于原始數據的導入和可視化。而后需評估各個樣本的質量,如果某樣本在全基因組上被成功分型的位點數目低于芯片全部預設位點數目的95%,則認為該樣本DNA發生降解,故而將該樣本剔除。在剩余樣本組成的群體中,再進行位點水平的質量評估,如果一個位點有超過3%的樣本沒有分型成功,分析該位點的統計學效力不足,這些位點也需剔除。
對通量較低的候選SNP位點進行分型,如熒光探針分型(TaqMan)、質譜分型(Agena Bioscience)、KASPar?等[26-28],受上樣量、檢測儀器、操作熟練程度等外部因素影響較大,分型成功率的閾值可降低到80%。
2.2.3 污染評估 可控和不可控因素引入的樣本污染,將降低來自污染樣本基因組學數據的可信度,因此污染樣本需要從研究群體中刪除。
樣本污染主要來自微生物和人源DNA。被微生物污染的樣本,測序得到的基因組GC含量明顯異于正常人類基因組GC含量(39%~45%),因此可以通過全基因組測序數據預處理、計算GC含量來判斷。人源DNA污染會導致被檢測樣本的基因組上存在過多的雜合變異,可使用VerifyBamID軟件判斷全基因組測序和芯片分型樣本人源污染情況[26]。2.3 群體遺傳學質控 無污染、遺傳變異檢測成功率合格、數據量滿足要求的樣本,可以按照研究目標的不同,開展面向群體的關聯分析、面向家系的連鎖分析,以及面向單基因病個體的突變致病性注釋等研究(圖1)。其中用于關聯分析、連鎖分析的樣本,需要進行群體遺傳學質控。對于關聯分析,研究群體中不得存在重復樣本、具有親緣關系的樣本或遺傳背景明顯離群的樣本,同時評估群體的亞分層等群體遺傳結構;對于連鎖分析,需通過群體遺傳質控確定家系中沒有樣本遺傳數據與親緣關系不匹配的情況。使用RelPair、KING等軟件[29-30],分析樣本間的親緣關系以及樣本有無重復。對于關聯分析,以保留盡可能多的樣本進入分析流程為原則,去掉具有親緣關系的樣本對中的一個樣本,確保納入分析的樣本中不存在具有3度以內的親緣關系樣本。
遺傳背景的離群值和人群亞分層使用主成分分析進行。在全基因組上選取沒有連鎖不平衡關系的SNP位點,使用GCTA軟件進行主成分分析[31],使用STRUCTURE軟件評估人群遺傳結構[32]。本分析流程使用千人基因組數據庫中的中國南北方漢族,或歐洲人群、非洲人群作為參考人群[33],對目標群體的樣本進行主成分分析和聚類分析。如果目標群體在前二或前三個主成分中有明顯的離群個體,或者明顯和其他族裔人群聚類接近,則將該樣本刪除。如果人群具有明顯的分類,需要在后續的關聯分析中,將該樣本的前2~10個主成分作為協變量,以降低人群亞分層對關聯分析結果的影響。
上述群體遺傳學質控只能使用覆蓋全基因組的SNP位點來進行,因此對于候選基因SNP分型的項目和樣本,無法進行相應的群體遺傳質控。
在CNSR-Ⅲ研究隊列全基因組測序樣本中,完成群體遺傳學質控后,最終獲得無3度以內親緣關系、遺傳背景離群和亞分層的研究人群,樣本共計10 241例,這些樣本可用于全基因組關聯分析。
2.4 變異功能注釋 注釋是根據遺傳變異在基因組上的物理位置、變異形式等信息,解析和預測該遺傳變異的生物學功能的流程。根據注釋方法和內容的不同,注釋在蛋白編碼和基因表達調控兩個水平上展開。
2.4.1 蛋白編碼區注釋 位于基因的編碼區遺傳變異會直接改變密碼子的組成,對蛋白編碼造成影響。同時,基因內含子區的可變剪切位點發生變異,會影響蛋白編碼中外顯子和開放閱讀框的選取,也會影響蛋白質中的氨基酸序列排布。選取VEP(ensembl variant effect predictor)和ANNOVAR(annotation of genetic variants)軟件[34-35],對基因編碼區的變異進行注釋,使用dbscSNV數據庫和SpliceAI軟件對可變剪切位點的變異功能進行注釋和預測[36-37]。
2.4.2 表達調控區注釋 基因組上超過97%的序列為非編碼區,但是這些序列可能執行調節基因表達的功能[38]。這些區域發生的DNA變異有可能改變轉錄調節因子的結合能力,從而影響基因表達。對于表達調控區變異的注釋,主要通過檢索RegulomeDB、GTEx、GeneHancer等數據庫來完成[20-22]。通過判斷遺傳變異是否位于表達調控區預測對轉錄調節因子結合能力的影響,判斷遺傳變異對基因表達的潛在影響。
2.4.3 變異致病性評估 對于單基因病患者的基因組研究,主要目標是確定導致患者發病的變異。因此對該類患者攜帶的遺傳變異,除了進行功能注釋,還需要進行致病性評估,在實踐中采用軟件評估和數據庫檢索兩種方案進行。軟件預測采用InterVar等軟件[39],按照美國醫學遺傳學與基因組學學會(American College of Medical Genetics and Genomics,ACMG)標準[40],進行變異的致病性評估。數據庫檢索是從ClinVar[23]、OMIM(https://omim.org)、Orphanet(www.orpha.net)等數據庫中檢索患者攜帶的遺傳變異是否為數據庫已收錄的致病變異,并分析患者表型與數據庫收錄的表型是否相符。
2.5 連鎖分析 連鎖分析是在疾病家系中,按遺傳多態和疾病表型的共分離關系,定位疾病致病變異的分析(圖1),該分析在具有親緣關系的家系樣本中展開。可以使用的軟件包括Merlin(multipoint engine for rapid likelihood inference)、Haplo2Ped等[41-42]。需要注意的是,連鎖分析通常是將遺傳變異作為標注基因組區段的分子標記,進行致病基因所在區段的定位,并不能直接定位致病突變。因此,須對連鎖分析得到的陽性區段內所有遺傳變異進行注釋和精細定位,并進行分子生物學、細胞生物學實驗,對可能致病的遺傳變異進行功能驗證,才能確定致病變異。
2.6 關聯分析 關聯分析是指以不同個體的遺傳變異信息作為自變量,個體的表型信息作為因變量,在群體層面研究遺傳變異和表型的相關關系的方法[43]。按照遺傳變異在群體中的頻率不同,分別進行常見變異關聯分析和低頻變異關聯分析。
2.6.1 常見變異關聯分析 常見變異是指次要等位基因頻率(minor allele frequency,MAF)在群體中>1%,或者在研究樣本中攜帶者數量超過一定閾值(如30個)的遺傳變異。常見變異關聯分析采用廣義線性模型對遺傳數據和性狀信息進行線性擬合,對于數量性狀和質量性狀,分別采用線性回歸和logistic回歸的方法。常見變異關聯分析使用PLINK軟件開展[44],以單獨一個遺傳變異位點為單位進行計算,估計其與表型的相關性。
在常見變異關聯分析的過程中,由于遺傳變異測序或分型失敗,導致一部分樣本的個別位點,或者樣本群體的部分位點基因型數據缺失。這時可采用基因型填補(impute)方法,推測缺失位點的基因型。該操作基于參考群體的基因組遺傳變異和數據缺失的遺傳變異之間連鎖不平衡關系,推斷基因型已知的遺傳變異位點周邊基因組區域中,基因型未知或未被分型的遺傳變異的基因型。該操作對于常見變異表現較好,但是能夠被推定基因型的遺傳變異數量有限。
在常見變異關聯分析中,基因型填補能夠增加基因組的覆蓋范圍和密度,提高用于關聯分析的遺傳變異數量。在本流程中,使用IMPUTE2軟件及其相應的參考人群數據,完成基因型填補過程[45]。
2.6.2 低頻變異關聯分析 低頻變異是指MAF<1%,或者在研究樣本中攜帶者數量低于一定閾值(如30個)的遺傳變異。盡管低頻變異的數量眾多,但從單個遺傳變異的頻率來看,攜帶者數量較少,因此不適宜采用常見變異關聯分析中,以單個變異為單位進行關聯分析的方法。而是將一段基因組區域內的全部低頻變異進行歸納,依據頻率等因素對每個低頻變異進行加權,構建統計量,再與表型進行關聯分析。在本流程中,使用SKAT、rvtests等軟件[46-47],以基因組區段為單位,進行低頻變異關聯分析。
2.6.3 薈萃分析 整合不同研究的全基因組關聯分析結果進行薈萃分析,可以提升樣本量,增加統計學效力。對于可以開展薈萃分析的項目和數據,使用METAL和RARE METAL軟件[48-49],進行常見變異薈萃分析和低頻變異薈萃分析。
2.7 跨組學研究 關聯分析得到的結論往往只能表明相關性而不是因果關系,因此關聯分析的研究結果難以實現臨床轉化。在大數據、多組學時代,可以通過跨組學分析的方法,證明因果關系或者中介因素。使用遺傳多態作為分組依據,模仿隨機對照試驗對樣本分組進行的孟德爾隨機研究,能夠推斷來自不同組學的表型數據之間的因果關系。為此,在流程中設計基于CIT包的因果推斷檢驗[50],以及基于TwoSampleMR包的孟德爾隨機分析[51]。
3 討論
腦血管病是嚴重威脅我國國民健康的復雜疾病,對腦血管病分子機制的研究,有助于發現潛在的藥物靶標,開發新的治療方案。以高通量測序和質譜為代表的組學技術蓬勃發展,推動了腦血管病在基因組、轉錄組、表觀遺傳組、蛋白質組、代謝組、宏基因組等領域研究的長足進步,加深了對腦血管病分子機制的認識。但是由于種種限制,以往研究樣本量較小、組學數據維度單一,通常只能證明相關性而非因果關系,因而對臨床治療的幫助有限。在大樣本量隊列中展開多組學分析,不僅能夠從多個維度揭示疾病分子機制,也能更清楚地確定生物標志物、藥物靶標和疾病結局的因果關系。多組學是融合了基因組、轉錄組、表觀遺傳組、蛋白質組、代謝組、宏基因組等多個組學的綜合性學科。一次性開展多個組學的研究,成本巨大而且風險較高,因此在多組學研究中需要進行良好的頂層設計和分析,以期為腦血管病的臨床治療和研究提供新的支持[52]。
以往生物信息學標準化分析流程的研究中,對于基因組數據的標準化分析關注較少[53]。以往腦血管病的基因組學研究,由于目標、組學技術和數據的差異,分析策略和流程的選擇較為單一。因此,基因組學數據的分析流程缺少有效的集成和優化,難以滿足腦血管病等多種復雜疾病的研究需求[54]。不同亞型的腦血管病臨床表現難以區分,單基因病和多因微效型腦血管病患者臨床表現相似,增加了分型診療的難度。對于腦血管病的大規模測序研究,建議先對單基因病型腦血管病患者進行致病性評估和連鎖分析,為臨床診斷定位可能的致病突變。在關聯分析中,優先保留多因微效型腦血管病患者的資料,增強樣本遺傳架構的一致性和研究的統計效力。為了分析CNSR-Ⅲ研究隊列基因組學數據,在調研研究需求和腦血管病特點的基礎上,搭建了基因組學數據分析流程。該流程以模塊化設計思路,包含了關聯分析、連鎖分析、遺傳變異功能注釋、致病性評估、跨組學分析等多種分析方法,能夠滿足腦血管病研究所需的單基因病患者評估、關聯分析等功能。可以根據不同的研究目的,有選擇地使用本流程的不同模塊。本流程為分析腦血管病基因組數據提供了全面、系統的方案。同時,本研究關注了以往研究論文較少提及的高通量組學數據的管理方案,介紹了組學數據接收、核對、安全管理的原則,有助于腦血管病領域基因組學和多組學研究的開展。
本研究建立的分析流程在以下方面需要繼續優化:首先,本流程主要用于基因組DNA遺傳變異的檢出和研究,對多組學包含的其他組學數據,如轉錄組、表觀遺傳組、蛋白質組、代謝組等研究產出的數據,尚未搭建分析流程。其次,本流程適合于基因組DNA發生的種系變異,不適用于體細胞變異的分析。在CNSR-Ⅲ研究后續的基因組等多組學研究中,隨著研究的深入和其他組學數據的產生,將逐步完善分析流程。最后,需要指出的是,盡管能夠通過流程,提升數據分析效率,但是在臨床科研的實際操作中,具體的參數設置,如協變量、病例-對照分組等,都會對分析結果產生影響。因此在分析過程中,仍然需要根據臨床和生物學知識,調試相應參數以獲取合理的分析結果。
說明:本文涉及的部分生物信息學術語或數據庫名稱在國內尚無統一譯文,強行將這些術語或名稱翻譯成中文將影響讀者對原意的理解,因此本文對此類術語及名稱未進行翻譯。
【點睛】標準化、模塊化生物信息分析流程的確立,為全面挖掘腦血管病基因組學數據、展示其蘊含的致病信息提供了系統性解決方案,將推動腦血管病多組學研究的發展。
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
電子制作(2018年18期)2018-11-14 01:48:24
汽車工程學報(2017年2期)2017-07-05 08:13:02
山東工業技術(2016年15期)2016-12-01 05:31:22
中國中醫藥現代遠程教育(2014年11期)2014-08-08 13:23:44
