二氧化碳轉化為合成氣及高附加值產品的研究進展
2022-04-12 03:54:26邵斌孫哲毅章云潘馮弘康趙開慶胡軍劉洪來
化工進展 2022年3期
關鍵詞:催化劑
邵斌,孫哲毅,章云,潘馮弘康,趙開慶,胡軍,劉洪來
(華東理工大學化學與分子工程學院,上海 200237)
煤、石油、天然氣等化石資源為現代社會發展提供了能源,但日益增長的CO排放已對人類和環境產生嚴重危害。2018 年,全球CO排放量約為42.1Gt,其中36.6Gt(87%)來源于以化石燃料為基礎的能源系統和化學工業。2020年大氣中CO濃度已高達416mL/m,是工業革命前的1.45倍。針對現有化石能源體系,碳捕集與封存(CCS)技術是減少CO排放的最有效途徑。目前全球CCS工業和示范項目已達到300多個,但總體規模遠不能滿足社會快速增長的需求。其主要原因在于CCS技術成本高,并且將捕集的CO封存的可持續性和安全性備受質疑。因此,發展碳捕集與利用(CCU)技術,將捕集的CO轉化高附加值的化學品和燃料,從而實現CO資源化利用,是一項重要的碳減排技術,對緩解能源危機和實現碳中和具有重要意義。
CO高效轉化技術常見產物有碳一化學品(CH、CO、甲醇等)、尿素、乙醇、聚酯、芳烴和烯烴等。CO分子的標準吉布斯自由能(Δ)為-394.38kJ/mol,其固有的熱力學穩定性與動力學惰性致使直接合成高附加值化學品效率低、反應條件苛刻、產物收率低。相對而言,CO轉化為CO是最切實可行的途徑,并且CO 具有高活性,將其與H按一定比例共混可以得到合成氣(CO+H),作為平臺分子進一步制取液體燃料、低碳烯烴、芳烴等大宗化工和能源高附加值化學品。目前,以CO為原料制備合成氣方法包括熱催化、電催化、光催化等轉化途徑,其中熱催化轉化具有處理量大、效率高、易于大規模工業化應用等優勢。相比較熱催化過程反應溫度高,電催化和光催化還原CO可以在常溫常壓下進行,也為工業上低能耗綠色生產合成氣提供了可行的技術路線。因此將CO熱催化、電催化、光催化轉化為合成氣以及合成氣進一步轉化高附加值化學品是實現CCU的重要途徑(圖1),其核心關鍵是設計和制備高效的催化劑。本文著重對比綜述了熱催化法、電催化法、光催化3種不同方法將CO高效轉化合成氣最新進展,總結了合成氣進一步轉化為高附加值化學品例如烯烴、液態燃料和芳烴等研究工作,分析了各反應過程催化劑設計方法,為CO高效轉化利用提供新的思路。
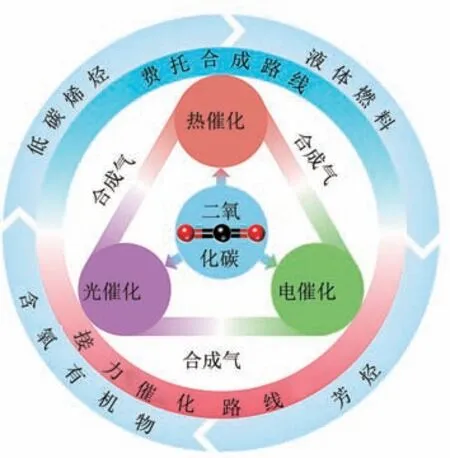
圖1 CO2轉化合成氣以及合成氣進一步轉化高附加值產品途徑
1 二氧化碳轉化為合成氣
將捕集的CO作為碳源轉化為合成氣,可實現碳循環利用,在解決環境問題的同時,協同突破資源與能源的碳中和的瓶頸問題。合成氣是化學工業生產中的一類重要原料。不同氫碳比(H/CO)的合成氣在化學合成中有著不同的應用。合成氣的生產主要采用天然氣催化重整和煤氣化工藝,高度依賴化石原料,造成了環境和能源的雙重壓力。因此,尋找綠色可替代的合成氣生產方法迫在眉睫。利用CO與甲烷或氫氣通過熱催化,可大規模生產合成氣,實現化石能源低碳利用;其次,CO電化學還原與水電解制氫耦合,在常溫常壓下將其成功轉化成不同氫碳比合成氣是一種清潔高效的生產方法;最后,利用清潔可再生太陽能通過光催化將CO還原為合成氣是一種綠色環保的新方法。
1.1 CO2熱催化制備合成氣
CO通過催化加氫制取合成氣,氫可由傳統意義的氫氣和富含氫的物質例如甲烷等低碳烷烴提供,通過包括二氧化碳-甲烷干重整(dry reforming of methane,DRM)和逆水煤氣變換反應(reverse water-gas shift reaction,RWGS)兩種典型反應過程實現。
1.1.1 二氧化碳甲烷干重整(DRM)制合成氣
DRM 主要通過CH催化還原CO[式(1)],可生產2倍體積的合成氣,是一種有效利用CO的方法。熱力學分析表明DRM是一個強吸熱反應,高溫有利于反應正向進行,但是過高的反應溫度對反應裝置要求較高,同時利于發生逆水煤氣副反應[式(2)],消耗產生的H,造成H/CO體積比下降。更值得注意的是所有合成氣生產過程都受到焦化的影響,由C-H-O 三元熱力學分析圖表明(圖2),550~700℃易發生的CO 歧化反應[Boudouar reaction,式(3)]和900℃以上CH的裂解反應[methane cracking,式(4)],都將導致催化劑表面積炭。因此,開發合適的催化劑對提高DRM 反應選擇性,抑制催化劑表面積炭意義重大。貴金屬催化劑例如Rh、Ru、Ir、Pt、Pd 等活性組分具有反應活性和選擇性高、反應溫度低、抗積炭和壽命長的特點,但其高成本限制了其大規模應用。而非貴金屬催化劑例如Ni、Co、Fe、Mo、Cu 等活性組分具有成本低和儲量來源豐富的優勢,其中Ni 基催化劑的活性和貴金屬催化劑相當,是近年來的研究熱點,但Ni 基催化劑在長時間高溫條件下容易失活,失活主要原因包括兩個:①CH裂解和CO歧化反應引起催化劑表面積炭失活;②金屬活性組分Ni高溫下燒結,使得催化表面活性點減少。

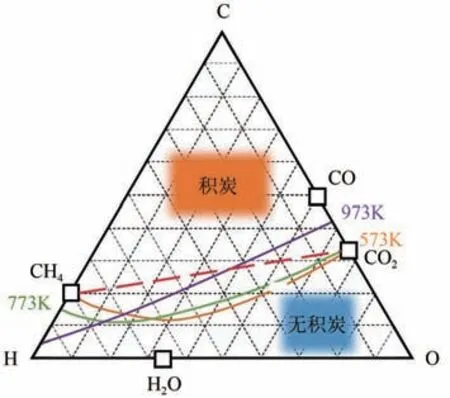
圖2 C-H-O三元相圖
反應機理的研究對于指導合成高效催化劑具有重要意義,DRM反應機理復雜,通常包含的主要步驟為:①CH和CO的解離或活化;②含有C、H和O元素的中間體在活性位點上的吸附;③表面反應和產物的生成;④CO、H和HO 等產物的脫附。Zhou等利用密度泛函理論(DFT)計算得出在活性Ni(111)表面的反應物、中間體和產物的吸附能以及涉及DRM 過程的可能進行的基元反應活化能(圖3),主要反應路徑包括CO直接解離出CO 和O,CH解離生成CH或C,CH 和C 被O 氧化,最后CHO 分解生成CO。Zhu 等建立了Ni 催化劑的微觀動力學模型,確定了3條反應速率接近的反應路徑,其中表面O 氧化C、OH 氧化C 和CH氧化反應路徑的貢獻率分別為73.1%、8.4%和18.5%,表明C 被表面O 氧化是主要反應途徑。因此,碳在催化劑表面沉積的速率和程度由CH解離、CO歧化和表面碳氧化共同決定。Zhang等報道揭示了Mo 在改善傳統Ni/Z 催化劑的催化性能和減緩積炭方面起促進作用,存在穩定、高含量的Ni,提供了更有效的甲酸鹽中間體的催化途徑以及反應過程獨特的MoO→ MoCO氧化還原循環,有利于積炭的去除。但由于DRM 反應過程每一步驟還包含多種可能性,目前真正的反應途徑和速率決定步驟(RDS)仍存在一定的爭議,有待結合催化劑特性深入探討。
圖3 CO2-CH4干重整體系在Ni基催化劑表面的正、逆反應在973.15K的反應能壘[16]
目前普遍認為催化劑的特性與顆粒大小、形貌、金屬活性位與載體間相互作用的強弱以及實驗條件等因素有關。金屬活性組分的尺寸超過臨界尺寸(10nm)時,才能滿足積炭成核的條件。但在DRM 高溫反應條件下,尺寸較小的催化顆粒普遍表現出較差的熱穩定性,容易燒結,因此制備高活性、高穩定性、抗積炭催化劑仍然是一個巨大的挑戰。Zhang 等設計制備了一種Ni 單原子型催化劑(Ni/HAP-Ce),通過與Ce摻雜的羥基磷灰石(HAP)相互作用,原子級分散的Ni 具有DRM 的高催化活性和抗積炭性能。實驗和計算研究表明,孤立的鎳原子具有只激活CH中的第1 個C-H 鍵的獨特性質,本質上避免了甲烷深度分解成積炭。由于金屬活性組分和載體之間存在普遍的相互作用,通過選擇合適的載體調控金屬-載體相互作用(MSI),可以提高Ni 在載體表面的分散度,一方面能夠提供更多的反應活性位點來提高反應活性,另一方面可以使Ni 納米顆粒的尺寸小于臨界尺寸,從而有效抑制積炭產生,增強催化劑穩定性。Yavuz 等報道了一種設計穩定高效甲烷干法重整催化劑的新策略——單晶邊緣納米催化劑(NOSCE)技術(圖4)。單晶MgO 載體的邊緣可穩定Mo 摻雜的Ni納米催化劑(Ni-Mo/MgO),其中Mo摻雜可以增強Ni 的氧化穩定性,促進Ni 顆粒向MgO 晶體的臺階邊緣移動。在DRM 反應中具有高效催化活性,并且連續運行850h 以上仍未失活,具有優異的抗結焦和抗燒結性能。通過摻雜另一種金屬形成Ni-M雙金屬合金催化劑也能有效提高熱催化DRM 的抗積炭性。Kim 等合成了粒徑恒定在5.5nm 左右的NiFe/MgAlO 雙金屬合金催化劑,與單一金屬催化劑相比表現出最佳的DRM 催化活性和穩定性。進一步揭示NiFe 合金表面上Fe 遷移形成了FeO 的關鍵作用:通過氧化還原機理恢復NiFe 合金并減少碳沉積。

圖4 Ni-Mo/MgO催化劑單晶邊緣負載納米催化劑(NOSCE)機制[22]
1.1.2 二氧化碳加氫(RWGS)制合成氣
將捕集的CO通過逆水煤氣變換[RWGS,式(2)]加氫(尤其是綠氫)還原為合成氣,將是未來形成封閉碳循環的重要一步。RWGS 是一個吸熱過程,高溫有利于反應,但往往伴隨著甲烷化副反應[Sabatier reaction,式(5)],導致CO產物的選擇性低。因此,開發高溫下具有高活性和選擇性的催化劑是關鍵。Kaiser 等分析了在H、CO摩爾比為3∶1的條件下RWGS 反應氣態產物的熱力學平衡組成(圖5)。當反應溫度低于600℃時,CH是主要產物;超過700℃時,CO產物明顯增多。但從降低能耗和成本角度分析,應避免高溫反應,因此需對RWGS反應機理深入理解,特別是結合實際催化劑探討反應路徑和反應動力學,從而獲得催化劑設計與制備規律,具有重要意義。
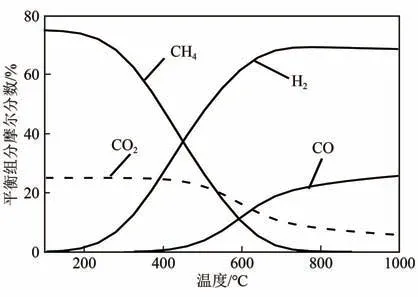
圖5 RWGS反應氣態產物的熱力學平衡組成
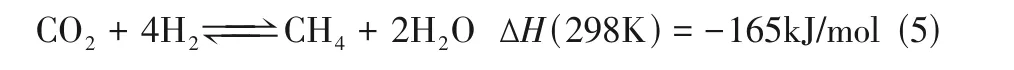
近年來,結合多種原位表征技術、同位素示蹤方法和基于DFT 計算對催化劑表面吸附物種演化的跟蹤,對RWGS 反應機理的研究取得了重大進展。表1列出了RWGS反應遵循的兩種典型反應路徑:①氧化還原機理,即CO在催化劑表面解離為CO和O,H與O反應,但并不直接參與CO的還原;②中間產物機理,即H與CO在催化劑表面直接反應生成中間體(如甲酸鹽、羧酸鹽或碳酸氫鹽等),然后進一步加氫生成CO和HO。氧化還原機理認為在金屬納米顆粒表面形成的CO 的結合強度可以決定產物的選擇性。CO與納米顆粒的強相互作用導致C—O鍵解離,有利于CH的形成,而CO與金屬的弱相互作用有利于CO 本身作為產品。因此,調控金屬表面對CO 吸附強度可以大幅提高逆水煤氣反應產物CO 的選擇性。中間體產物機理認為,中間物只在載體與金屬納米粒子的界面上形成,金屬納米粒子上吸附的氫原子可以與中間產物反應,以CO和HO的形式解吸。Szanyi等報道了兩種Pd/AlO負載型催化劑,基于原位紅外-質譜表征發現CO首先與氧化鋁上的羥基結合生成碳酸氫鹽,Pd上吸附的H與之反應生成甲酸鹽中間體。由于Pd有優先生成CO或CH的兩組不同晶面,與CO弱相互作用的Pd位點解吸產生CO,與CO強相互作用的Pd位點,CO進一步加氫生成CH。
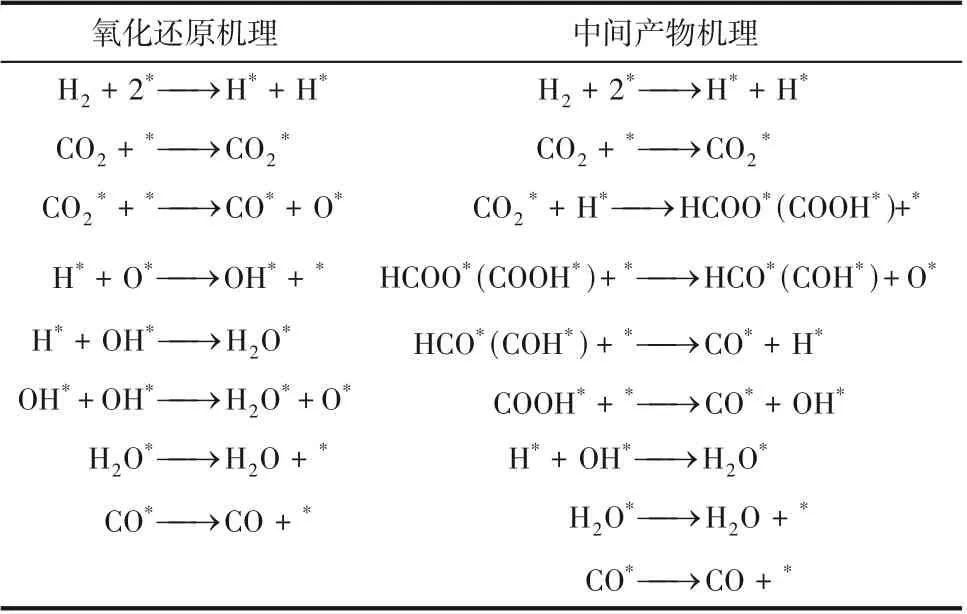
表1 RWGS反應的氧化還原機理和中間產物機理[25]
同時,催化劑表面的空位也被認為有助于CO的吸附和活化。因此,氧空位濃度高、金屬-中間物種鍵能強度弱的催化劑對RWGS反應具有較好的催化效果。最近Willauer 等開發了一種低成本、高活性和高選擇性的K-MoC/g-AlO催化劑,450℃時CO轉化率為44%,CO 選擇性為98%,運行10 天內沒有任何失活跡象。通過DFT 計算進一步發現反應過程中有更多氧覆蓋的MoC表面對CO吸附增強,容易加氫轉化為CH,會導致CO 選擇性下降。Hu等針對乙烯工業過程裂解高溫煙氣,將鈣循環(calcium-looping,CaL)和RWGS 反應相結合,通過設計氧化還原異質結的Fe/Co 合金催化劑(圖6),在同一固定床中成功實現了長周期高溫CO捕集和原位轉化,CO轉化率高達90%,CO選擇性100%。
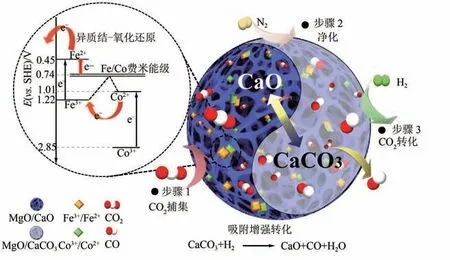
圖6 雙金屬催化劑Fe5Co5Mg10CaO異質結-氧化還原機理[30]
1.1.3 CO熱催化制合成氣的工業化進展
以二氧化碳規模化利用技術為核心的工業化碳負排方案中,CO-CH通過DRM 制合成氣是一種綠色的生產方式。目前天然氣除作為燃料外,約15%用作化工原料,假設其中30%用于CO-CH干重整并耦合合成氣直接制烯烴,則各類烯烴產量可達740 萬噸,同時直接消耗CO約1020 萬噸。2010年12月,美國Carbon Sciences Inc.公司與加拿大薩斯喀徹溫大學(UOS)就DRM 催化劑技術簽署了全球獨家許可協議。在反應器連續2000h的實驗中,UOS開發的催化劑轉化率達到92%,無明顯的燒結和積炭現象。國內上海高等研究院孫予罕教授團隊在成功解決Ni-CaO-ZrO納米復合催化劑的抗焦化問題基礎上,完成了催化劑研制和反應器模擬研究以及百噸級催化劑的工程放大和生產,并于2017 年在山西潞安集團建成了全球首套萬噸級CO自熱重整制合成氣中試裝置,日轉化利用CO高達60t,合成氣產量高達20m/d。該CO-CH干重整制備合成氣技術的成本為500~600CNY/t 合成氣,與煤制合成氣技術的成本相當,相較于目前傳統的水蒸氣重整生產合成氣,其成本可以降低20%。實際DRM工業化應用過程中仍需解決的問題包括:①高壓操作的必要性,尤其是生產得到的高壓合成氣對于進一步轉化為其他產品具有優勢;②調控合成氣中碳氫比,獲得目標合成氣。
雖然RWGS反應是活化CO的重要的一步,但目前還未見CO轉化利用的工業開發應用。Rezaei等通過Aspen 模擬分別評價了RWGS 工藝和DMR 工藝兩種不同合成氣生產方式的經濟性。與RWGS工藝相比,DMR 工藝生產H/CO 比為1 和2 的合成氣的總成本更低。以H價格、CH價格和碳稅進行盈虧平衡分析發現當H價格為1000USD/t,CH價格和碳稅分別為740USD/t 和44USD/t 時,RWGS 工藝與DMR 工藝相當。目前的研究重點是降低H生產成本和強化RWGS反應,從而降低工業應用RWGS過程的總成本。但是RWGS工藝作為合成高附加值產品串聯反應的第1步,在CAMERE甲醇合成工藝中得到了實際工業應用。同時,RWGS過程與烷烴脫氫耦合是工業化高效生產烯烴和合成氣的途徑。因此,通過RWGS 反應將CO轉化為高附加值化學品和燃料具有廣闊前景。
1.2 CO2電催化轉化合成氣
相比較于熱催化轉化CO,電催化還原CO的方法具有催化效率高、操作條件簡單和反應條件溫和等特點。目前大部分已報道的電催化劑都集中于CO高選擇性催化轉化為CO。本節通過概述最新的高效CO電催化轉化合成氣的催化劑研究進展,探討其工業化應用前景。
1.2.1 電催化還原CO基本原理
CO電催化還原為CO的反應機理主要包括3個步驟:①CO的活化生成CO;②經過質子-電子轉移(PET)先生成COOH中間體,再進一步生成CO和HO;③吸附在催化劑表面的CO脫附。由于析氫反應電位低,是溶液體系CO電催化還原不可避免的副反應。由反應機理分析可知,相應的高效電催化劑應滿足:①對CO具有較強的吸附活性和還原能力;②對關鍵中間體(COOH)具有適當的吸附強度;③CO吸附能適中、還原能力弱。因此,開發性能優異、合成方法簡單、來源豐富和低價的CO電還原催化劑是關鍵。
1.2.2 電催化新材料
目前高效CO電催化劑主要包括納米單金屬催化劑、納米合金催化劑以及過渡金屬單原子催化劑等。建立電催化劑微觀幾何結構與電子分布的調控方法,探索微觀結構與催化性能的關系是研究的重點。微觀幾何結構中催化劑的尺寸效應尤為顯著。Liu等通過DFT計算發現,當Cu納米顆粒降低至由309個原子組成時呈現顯著的尺寸效應。當顆粒由147 個原子組成時,CO電催化轉化為CO過程具有最合適的COOH吸附能以及CO脫附能,對應最低的CO 生成能壘,同時也能抑制H的形成。類似的尺寸效應在超細Au 納米顆粒也有體現(圖7)。
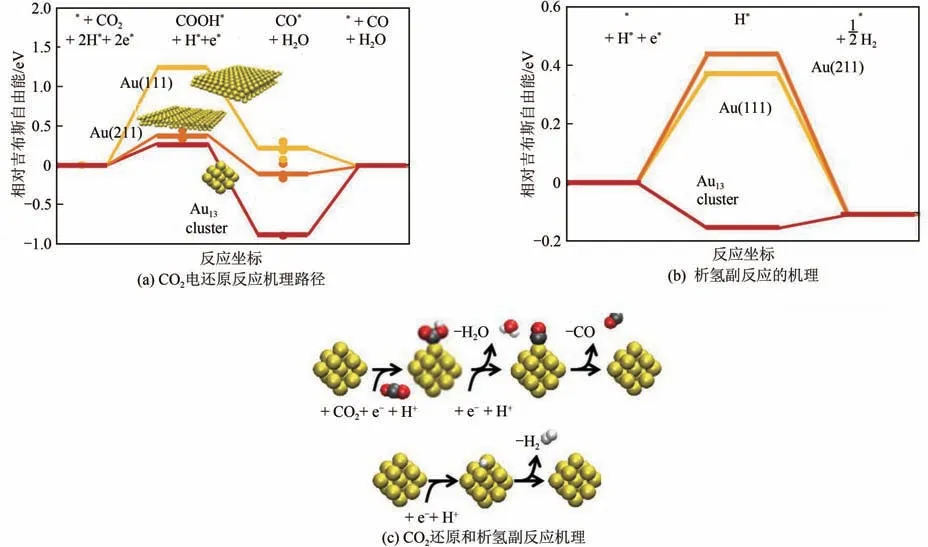
圖7 Au納米顆粒尺寸效應對電催化還原CO2機理的影響[39]
進一步將催化劑金屬活性組分尺寸降低到原子尺度,金屬單原子電催化劑呈現出不同尋常的CO電催化還原活性,并且金屬與載體之間的MSI相互作用更為顯著。這種效應往往能夠顯著調控電催化反應中各個中間物種的吸附能,從而提高反應的活性。因此,單原子金屬電催化劑的原子利用率極高,催化位點明確,易于剖析催化反應路徑等優點。其中Ni單原子催化劑表現出極佳CO電催化還原性能,CO 的法拉第效率幾乎可達到100%。Gong 等利用MOF 中低沸點金屬Mg,通過熱解法控制Ni 單原子的N 配位數Ni-N-C(=2,3,4),其中Ni-N-C 的CO 法拉第效率最高達到97%,對應的轉化頻率(TOF)高達1622h。此外,其他過渡金屬單原子電催化劑如Fe、Co 等,均能夠通過適當的合成方法精確調控配位環境從而實現CO高效轉化為合成氣。載體的性質也會對催化活性起到至關重要的作用,Zhu 等通過DFT 計算發現石墨烯上的Fe-N位點對CO的吸附能過強而易被毒化,而富缺陷的石墨烯則能夠促進Fe-N位點上CO 的脫附從而提高CO的法拉第效率。
通過優選雙(多)金屬摻雜組成和配比精準調控催化活性位點d帶電子結構的策略,也廣泛應用于提高CO電催化效率,調控合成氣的碳氫比。Han 等報道了一種Cu-Co 合金電催化劑,其在含離子液體的甲腈電解液中CO 的法拉第效率最高可97.4%,Cu和Co的比例決定了合成氣中H和CO的比例。此外,He等也發現Co-N位點能夠高效析氫而Ni-N位點能夠高效產出CO,兩者的共存能夠實現合成氣比例的精準調控。
結合金屬單原子催化效率和雙金屬催化的調控性,金屬雙原子對電催化劑協同促進催化的現象引起了關注。Zhao等制備了含有豐富Ni-Fe雙原子對位點的電催化劑,在很寬的電勢范圍(-0.4~-1.0V)內均能高效產出比例為8∶1的合成氣,CO的最優法拉第效率高達98%。DFT計算進一步揭示了這種獨特Ni-Fe 雙原子對可以使CO在兩個金屬位點交替協同促進轉化為CO(圖8)。
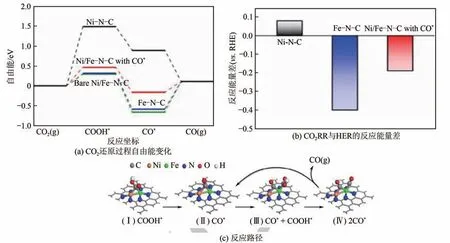
圖8 Fe-Ni雙原子對位點上CO2電催化還原的機理[47]
1.2.3 CO電催化制合成氣的工業化進展
近年來電催化反應體系從實驗室小試、中試示范到工業應用不斷發展,電極從最初的玻碳電極變為氣體擴散電極;電解池類型從H型電解池到流動池,再到兼具兩者優點的膜電極體系(MEA),目前工業化的CO電解電流密度可在0~1000mA/cm范圍內變化,對應的合成氣比例在(4∶1)~(1∶1)靈活調控。
近日,中國石油和化學工業聯合會在內蒙古對CO電解制合成氣中試裝置進行了現場考核。此裝置自從2020年8月開車以來,一直保持穩定運行狀態,其能夠穩定產出碳氫比為0.52∶1 的合成氣,直流電耗電量為6.69kW·h/m。此裝置每年可處理30t CO,生產45000m合成氣,副產22500mO。因此,CO電催化還原生產合成氣是極具前景的節能減排策略。
據CO電催化還原成本分析(圖9),目前電催化還原CO制CO 成本為130USD/t,僅次于甲酸(80USD/t)。但是液態的甲酸需從電解液中分離提純,其對應的成本高達60USD/t。而氣態CO 可采用簡單的變壓吸附進行分離提純,對應成本則僅為10USD/t。因此,CO電催化制合成氣最具經濟性,并且合成氣可以作為重要化工平臺氣原料,制備系列的精細化學品,進一步提高其附加值;同時在技術層面最易實現大規模電解技術。

圖9 CO2電催化還原制備各種產物的經濟效益分析[52]
但目前工業化電解CO制合成氣還面臨著巨大的挑戰,主要體現于:①工業化過程需施加較高的電壓提高反應速率,確保連續規模化生產量。如何在高電流密度下(>200mA/cm)提高CO 產物的選擇性,從而避免后續產物的分離是重要問題。②在電催化過程中,二氧化碳還原(CORR)和水還原(HER)反應具有競爭關系,電解過程既要保持兩個反應的活性,又要同時實現碳氫比可調的合成氣,具有一定的挑戰性。③陰極CO電催化還原效率還會受到陽極半反應(通常為析氧反應)的影響,整個電解體系中陽極上的電能損耗可高達90%。如何提高電解整體能源效率,需兼顧解決電催化析氧反應的動力學制約問題。
1.3 CO2光催化轉化合成氣
光催化技術利用清潔可再生太陽能轉化為化學能,兼顧能源、環境和經濟要求,是最具前景的綠色轉化技術。本節聚焦光催化CO制合成氣的機理,通過概述光催化最新材料及改性策略等研究動態,以期探討規模化工業生產前景。
1.3.1 CO光催化轉化機理
CO光催化轉化是一個典型的多電子轉移過程,包括3個步驟:光催化劑對光的吸收;光生載流子的產生、分離和傳輸以及光生載流子和反應物之間的化學過程[圖10(a)]。光合作用是最經典的CO光催化原理,通過葉綠體,利用光能將CO和HO轉化成儲存著能量的有機物,并且釋放出氧氣的過程。光合作用過程至少包括一個光能轉換的葉綠素分子(P)、一個原初電子受體(A)和一個原初電子供體(D),才能將光能轉換為化學能,CO得到原初電子受體A提供的電子而還原為葡萄糖等有機物。光合作用過程中CO并沒有直接參與原初電子傳遞,其原因在于CO穩定性高、還原電位高,與葉綠素的帶隙能不匹配。為了尋找高效CO還原光催化劑,分析對比常見的半導體光催化劑的帶隙能與CO不同還原產物的電極電位,如圖10(b)所示,光催化劑的導帶需高于-0.52eV,才能將CO還原為CO。同時由于析氫反應的還原電位略低于CO/CO電位,因此析氫反應在CO轉化制合成氣過程中不可避免。要提高CO的光催化效率,光催化劑的結構調控是關鍵。
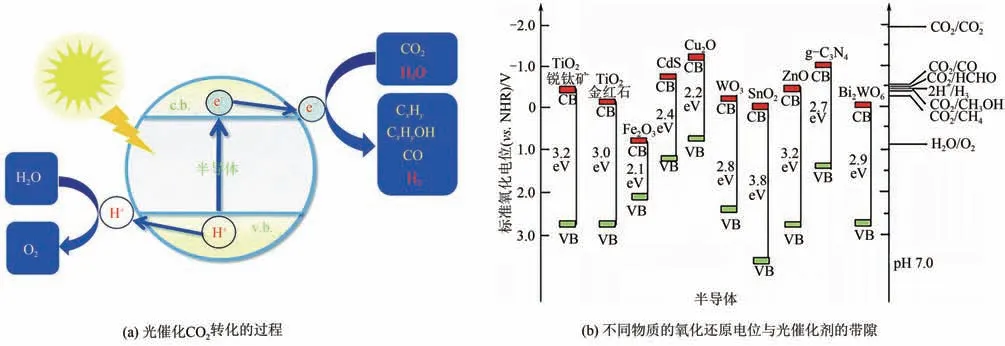
圖10 光催化CO2轉化的過程和不同物質的氧化還原電位與光催化劑的帶隙[57-58]
1.3.2 多孔光催化新材料
對于氣固光催化過程,CO氣體吸附在固體光催化劑表面是首要步驟。多孔光催化材料例如多孔聚合(POP)、金屬有機框架材料(MOF)、共價有機框架材料(COF)等新材料兼具CO吸附富集和光催化轉化雙功能,越來越受到關注。
POPs 是一類新型的結構多樣的高比表面聚合物材料,通過結構單元的設計與調控,強可見光吸收活性、光電性能可調以及高化學和熱穩定性等優點。近期,Yang等開發了曙紅Y基共軛多孔聚合物,在無任何光敏化劑或犧牲劑存在下,能有效地將CO和HO光催化還原為合成氣。
MOFs 中存在的開放金屬位點既可以作為CO分子的吸附位點,也可以作為CO光催化還原活性位點,各種MOFs 材料如HKUST-1、MIL-101、MAF-X27-OH、MOF-525-Co、NNU-31-M 等已成功應用于CO光催化還原。目前,MOFs 金屬節點物種、配體結構及其形成的晶體框架結構與光催化性能的關系是研究的重點。Dong 等通過調節MOF(PCN-250-Fe3)金屬簇節點中Fe的價態,實現了CO轉化活性的大幅提高,同時在光催化反應系統中可以檢測到H產物生成,形成合成氣。
COFs材料中存在大量π共軛結構,可使電子離域,已被用作電荷載流子傳輸的優良介質和光催化劑,并且COFs 表現出優異的CO吸附能力,近年來,D-A基COFs的探索也受到了廣泛關注。Lu等構筑了新型卟啉-四硫富瓦烯COF,缺電子金屬卟啉(TAPP)絡合物和富電子四硫富瓦烯(TTF)的鏈接增強了電荷分離效率、改善了光吸收,使CO高選擇性還原為CO。Zou 等合成了2,2’-聯吡啶基COF,與Ni 配位得到單原子分散(Ni-TpBpy),在將CO光催化還原為CO 同時產生H獲得合成氣(圖11)。
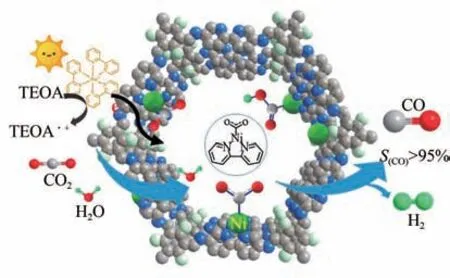
圖11 Ni-TpBpy光催化選擇性還原CO2[64]
1.3.3 光催化劑的改性策略
光催化還原CO和HO 被認為是一種綠色和可持續的合成氣生產方法。然而,該光催化反應的效率、合成氣中碳氫比的調控還存在較大的挑戰。最新的光催化改性策略聚焦于異質結的構筑、空缺位缺陷的調控等,以促進與調控CO還原反應(CORR)和析氫反應(HER)。
合理構建異質結構能夠有效提高光催化劑內部載流子的分離效率,并提升氧化還原能力。Yu等將Co 活性中心與三嗪框架(CTF)配位,形成分子內異質結,光吸收明顯增強,能夠在10h內產生3303μmol/g 的合成氣(CO∶H=1.4∶1),產率是不含異質結的CTF 的3 倍。Song 等通過靜電自組裝形成系列層狀雙氫氧化物/MoS異質結構的復合光催化劑,在可見光照射下將CO光還原得到的合成氣,碳氫比從(1∶1)~(9∶1)可調(圖12)。最近,Hu和Li等報道了一種環境穩定型黑磷異質結復合材料Pt/BP-OvMBWO,用于光催化CO還原制備合成氣[CO∶H=(1∶1)~(2∶1)],CO 和H的最大生成速率分別高達20.5μmol/(g·h)和16.8μmol/(g·h),且具有優異的循環使用穩定性。
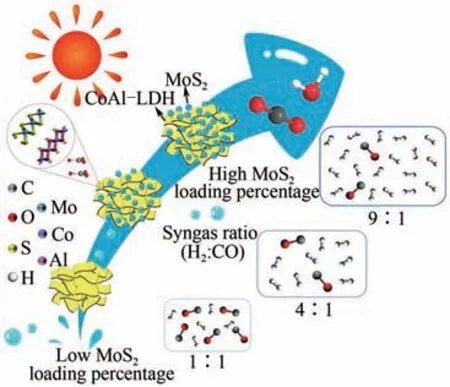
圖12 LDH/MoS2納米復合物通過CO2光還原合成可調諧合成氣[66]
將光敏劑與催化劑偶聯也是構筑異質結最常用方法。Zheng 等以超薄的層狀雙氫氧化物(LDH)為載體制備了高度分散Pd 的催化劑,與Ru 絡合物敏化劑配合形成高效異質結,在可見光照射下得到的合成氣碳氫比在(1∶0.74)~(1∶3)可調。Wu 等在氮摻雜碳上構建Mn 單原子催化劑MnSAs。以[Ru(bpy)]Cl作為光敏劑,以三乙醇胺作為犧牲劑,光催化CO還原制合成氣。CO和H的產氣速率分別高達1470μmol/(g·h)和1310μmol/(g·h),碳氫比從1.12~0.43 可調。同時,光催化劑的缺陷通常可以產生更多的電子空位,以強化電子空穴的分離,并增加CO的吸附。Wang等通過構建富含S空位的VS-ZnInS納米片,強化光吸收、電子-空穴分離和CO吸附,同步促進CORR 和HER 反應,CO∶H比為1∶1。與純ZnInS相比,合成氣產率提高了約4.73倍。Yang等報道了一種在聚合氮化碳(PCN)光催化劑表面生成氮空位(NV)的方法,以加速PCN光生載流子的分離和轉移動力學。在可見光下,NVs-PCN 的合成氣產率幾乎是原PCN的10倍。重要的是,通過調節NVs的濃度,合成氣H∶CO比可以在(0.24∶1)~(6.8∶1)之間調節。
1.3.4 光催化制合成氣規模化研究的挑戰
目前,光催化還原CO的研究主要集中在實驗室規模,距離規模化實際應用還面臨諸多問題,例如:①對CO光催化還原的機理,包括具體中間體、反應途徑和反應動力學認識不足,對提高CO光催化還原效率還處于“試錯”階段。②實驗室研究將固體光催化劑粉末直接加入CO溶液中,以充分提高光吸收、氣固兩相接觸和光催化效率。但是懸浮體系伴隨著光催化劑的后續分離和回收難度大、重復利用效率低等問題。③目前CO光還原產物主要集中于C化合物,通過光催化還原CO獲得高附加值產物仍面臨挑戰。因此,開發具有高活性、高反應選擇性和穩定性的新型光催化劑迫在眉睫。合理設計高效的無貴金屬助催化劑,有助于構建低成本。高效的光反應器和反應系統工程對于實現高效的CO減排至關重要。
光催化劑成型工程技術研究是工業化應用的首要關鍵,催化劑固定化避免了光催化劑分離問題,是一個值得關注的新方向。采用氣固相反應技術可實現高效氣固光催化,避免水溶液懸浮體系中CO氣體溶解度低、光催化劑相容性差、負載型顆粒易脫落等穩定性問題。通過反應器設計改進紫外光源穿透模式,可以較大程度提高光催化效率,使光催化制合成氣具有潛在的工業化應用前景。
2 合成氣制高附加值產品
CO通過熱催化、電催化和光催化過程可高效轉化為合成氣,合成氣作為生產合成氨、羰基合成醇、費托合成的原料平臺,可進一步轉化成高附加值化學產品和燃料,從而提升整個CO減排過程的可行性和經濟性,具有重要的研究意義。本節聚焦合成氣制低碳烯烴和芳烴的最新進展。
2.1 合成氣制低碳烯烴和液體燃料
合成氣制長鏈烴的催化過程為經典的費托合成(Fischer-Tropsch synthesis,FTS)。如圖13 所示,其主要反應機理包括:首先CO和H分子在催化劑表面發生解離吸附;然后CO 解離形成的C 物種加氫形成CH中間體(0≤≤3);這些中間體會進一步發生C—C偶聯生成CH中間體或者生成CH;最后CH經加氫或脫氫形成不同碳數的烷烴和烯烴。因為費托合成遵循表面偶聯機理,產物服從Anderson-Shulz-Flory(ASF)分布,其中產物C~C烴類的選擇性不超過58%,汽油(C~C)組分選擇性不超過48%。因此,如何將合成氣高選擇性地定向轉化為特定的高附加值化學品具有重大挑戰。
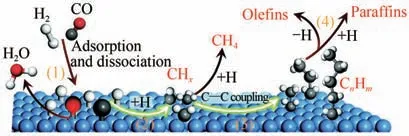
圖13 費托合成反應機理中關鍵步驟[72]
目前費托合成的研究主要集中于金屬氧化物及其碳化物型催化劑,例如Fe、Co和Ru等。Sun等研制出棱鏡型的CoC催化劑,可以在250℃、0.1MPa溫和的條件下高選擇性地將合成氣催化轉化為輕質烯烴,選擇性為60.8%,而甲烷選擇性低至5.0%。近期,Ding等發展了一種新型疏水性FeMn@Si催化劑,在模擬工業反應條件下(320℃和2~3MPa),實現了合成氣高選擇性制取烯烴,烯烴產率高達36%,選擇性可達65%,CH和CO等C副產物選擇性顯著降低至22.5%。進一步研究發現,FeMn@Si表面的SiO疏水殼層可保護碳化鐵活性相免受水的氧化,并保持碳化鐵活性相處于良好的穩定狀態。Ma 等開發出Na 修飾的FeC-ZnO 催化劑,在340℃、2MPa的條件下烯烴選擇性高達78%,其中主要以高碳-烯烴為主,烯烴的時空收率超過4000mg/(g·h),同時甲烷和CO的選擇性分別控制在8%和25%。
包信和院士團隊提出了“OX-ZEO”一步法制取低碳烯烴技術路線,如圖14 所示,其反應機理為合成氣先在金屬氧化物上反應生成乙烯酮(CHCO)中間體,該中間體進入鄰近的分子篩孔道,在分子篩擇形酸性位催化作用下生成低碳烯烴。他們采用ZnCrO金屬氧化物和介孔SAPO-34(MSAPO)分子篩組成雙功能催化劑,具有優異的低碳烯烴選擇性,在400℃、2.5MPa、合成氣H/CO為1.5 反應條件下,低碳烯烴選擇性高達80%,突破了ASF 分布,但CO 轉化率較低為17%。基于此反應,2019年9月,中國科學院大連物理化學研究所與陜西延長石油集團合作,成功完成煤經合成氣直接制低碳烯烴技術工業試驗,CO 單程轉化率超過50%,低碳烯烴選擇性優于75%,總體催化性能優于實驗室水平,進一步驗證了該技術路線的先進性和可行性。最近,包信和院士團隊通過對比研究了ZnCrO-SAPO-34 和MnO-SAPO-34 兩種雙功能催化劑,進一步揭示了金屬遷移的影響,Zn物種易于遷移至SAPO-34 上,形成Zn-OH,屏蔽了分子篩的Br?nsted酸中心,導致低碳烯烴選擇性下降。相比之下,Mn物種不會發生金屬遷移現象,在納米尺度上MnO與SAPO-34 兩種催化活性位越近,越有利于乙烯酮中間物擴散,低碳烯烴選擇性越高,這為進一步開發該類雙功能催化劑提供了基礎。Wang 等開發了由ZnO-ZrO雙金屬氧化物和SAPO-34 分子篩組成的雙功能催化劑。在400℃、1MPa、H/CO=2反應條件下,低碳烯烴選擇性高達74%,但CO 轉化率為11%。他們通過改進分子篩結構得到ZnO-ZrO/SSZ-13 的雙功能催化劑,將CO轉化率提升至29%,同時低碳烯烴選擇性77%。機理研究表明,CO首先在ZnO-ZrO表面轉化為甲酸類化合物,然后甲酸類化合物通過氫化反應轉化為甲氧基化合物。最后甲氧基化合物經分子篩進一步催化為烯烴。
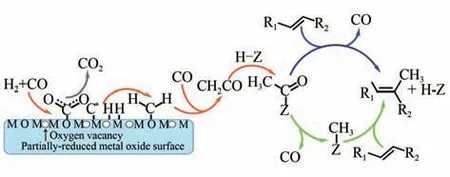
圖14 在ZnCrOx/SAPO-34催化劑上合成氣經乙烯酮中間體轉化為低碳烯烴的反應機理[72]
此外,合成氣也可以通過費托合成與加氫裂解/異構化反應耦合而成的接力催化方式高選擇性地轉化為液體燃料如汽油和異構烷烴等。Sun 等通過無溶劑研磨法制備出弱酸性Silicalite-1 分子篩封裝金屬鈷催化劑。在CO 轉化率約為30%的情況下,汽油選擇性高達70%,異構烷烴選擇性可達30.7%。包信和院士團隊開發出ZnMnO/SAPO-11 雙功能催化劑,該催化劑可以直接選擇性將合成氣轉化為高品質汽油。在CO 轉化率為20.3%時,汽油在烴類中的選擇性高達76.7%,CH的選擇性僅為2.3%。劉中民院士團隊構建出CuZnAl 與AlO混合組分和ZSM-5組成的雙功能催化劑,實現了合成氣向汽油的高效轉化,CO 單程轉化率高達86.3%,除開CO后C~C汽油產物選擇性高達80.6%,并且催化劑在110h的催化測試中催化性能保持穩定。此外,C含氧化合物特別是乙醇可用作燃料添加劑和綠色有機溶劑。Wang 等設計出由K-ZnO-ZrO、H-MOR 和Pt-Sn/SiC 組成的三步接力催化體系,通過將合成氣制甲醇、甲醇羰基化制乙酸、乙酸加氫制乙醇3個反應按接力催化的方式進行高效集成,實現了合成氣一步高選擇性制乙醇。乙醇選擇性可達70%~90%,突破了傳統過程的乙醇選擇性極限值。
2.2 合成氣制芳烴
通常合成氣制芳烴路線分為兩步:先把合成氣轉化為甲醇,再通過甲醇芳構化反應得到芳烴。但兩步法工藝繁瑣且甲醇制芳烴催化劑積炭失活嚴重。并且由于受費托合成反應產物ASF 分布限制,產物選擇性低。近年來,接力催化策略(OXZEO)在合成氣轉化中展現出獨特的優勢:通過反應的耦合不僅強化了反應過程,還保證了目標產物的高選擇性合成。因此,將合成氣制甲醇催化劑與H-ZSM-5 耦合,實現了合成氣直接高選擇性制芳烴。Ma 等將高效制備-烯烴的Na-Zn-FeC與改性處理后的介孔H-ZSM-5 分子篩混合,在340℃、2MPa 條件下實現了CO 轉化率為89%,芳烴選擇性為51%,時空收率可以達到16.8g/(g·h),同時CH和CO選擇性分別抑制在10%和27%的優異性能。Wang 等將ZnO-ZrO雙金屬氧化物和H-ZSM-5 分子篩組成雙功能催化劑,在400℃、3.0MPa、H/CO=2 的反應條件下,CO 轉化率可達20%,芳烴選擇性高達80%,甲烷選擇性抑制低于3%。更為重要的是該雙功能催化劑在1000h 反應過程中仍可保持穩定。最近,謝在庫院士團隊將CrO定向分布于ZSM-5 分子篩的(100)和(101)特定晶面得到高效雙功能催化劑,因其CO加氫形成的C中間體易擴散至鄰近的ZSM-5分子篩孔道內酸性位點發生芳構化反應,在395℃、7.0MPa、H/CO=1、1L/(g·h)反應條件下,CO轉化率高達49.4%。
但是合成氣制芳烴雙功能催化劑也存在一些問題:目前報道的CO 單程轉化率大部分不超過50%,較傳統的費托合成催化劑反應活性普遍偏低。因此需要進一步開發出高活性的多金屬氧化物來提高催化劑的反應活性。此外,接力催化反應條件一般發生在400℃左右,升高溫度對水煤氣反應(WGS)有促進作用,導致產生CO副產物,選擇性甚至高達43%。未來還需要進行更深的反應機理以及動力學研究,從催化原理本質上抑制WGS反應發生。
3 結語與展望
將CO高效轉換為合成氣,進一步通過費托合成路線或接力催化路線轉化為高附加值化學品和燃料成為發展高效利用非油基資源的新途徑(圖1)。基于熱催化、電催化和光催化這3 種不同方法以CO為原料制備合成氣的研究中已經取得了顯著進展,但距離大規模的工業化應用仍存在較大挑戰,開發出高效、高活性、低成本且穩定的催化劑是各技術推廣應用的關鍵。本文對比和總結了目前熱催化、電催化、光催化3種利用CO制備合成氣技術,見表2。其中應用成熟度最高的是加氫技術和甲烷干重整等熱催化技術,但反應條件苛刻、過程能耗大和催化劑穩定性等問題仍亟待解決。未來需要不斷完善催化劑活性位識別-結構-性能三者關系,實現耐高溫抗積炭高效催化劑的規模化制備;其次,通過整體系統的內部熱能利用和系統強化或耦合外部新能源系統供能,解決并實現反應器優化及其與反應過程的能量匹配;最后,利用合成氣作為后續化工生產的基礎原料制備高附加值化學品,形成完整集成工藝產業鏈,實現二氧化碳熱催化制備合成氣的規模化應用是其重要方向。

表2 CO2熱催化、電催化和光催化三種技術方法制備合成氣比較
隨著光伏、風電等綠色電源的發展,光、電催化將作為一種更加綠色高效的技術用于CO轉化制備合成氣。由于電催化劑在合成氣生產過程中容易出現CO中毒、高壓下H氣泡的抑制和活性位點的減少等失活問題,限制了該技術的大規模應用,因此未來在設計高效合成氣生產催化劑的過程中應考慮以下方面:①在催化劑表面形成多孔結構,促進氣體擴散;②優化催化材料的表面親疏水性,平衡水和CO的吸附;③合理設計催化劑獨特的多相界面,防止CO 中毒,增加反應活性位點數量。直接利用太陽能和地球上豐富的原料(HO、CO)光催化生產合成氣為CO資源化以及綠色能源利用提供了全新途徑。光催化劑活性主要受到電荷分離和轉移的制約,合理設計和制造高效且穩定的光催化劑是光催化CO產合成氣過程的核心。通過元素摻雜、缺陷工程、貴金屬負載和異質結構筑等策略,可顯著提高電荷載流子分離效率。基于光催化過程電子和質子轉移途徑以及化學鍵的形成/斷裂等機制的深入剖析,突破性提高光催化劑穩定性、轉化率和產率是光催化規模化應用方面的關鍵。
本文重點闡述了CO高效轉化為合成氣的途徑,未來將CO捕集與轉化過程耦合是CCU研究的重要研究方向,通過設計開發兼具CO吸附捕集與CO催化轉化的雙功能材料,有望實現CO高效捕集與原位轉化成高附加值產品一體化技術。充分提高能源利用率、有效降低減排成本,解決CO捕獲后提純和運輸的高額費用以及埋存所帶來的安全隱患,從而提升整體CCU技術的可行性和經濟性。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
