電催化還原CO2生成多種產物催化劑研究進展
2022-04-12 03:54:34鄭元波張前石堅李佳霖梅蘇寧余秦偉楊建明呂劍
化工進展 2022年3期
關鍵詞:催化劑
鄭元波,張前,石堅,李佳霖,梅蘇寧,余秦偉,楊建明,呂劍
(西安近代化學研究所,氟氮化工資源高效開發與利用國家重點實驗室,陜西 西安 710065)
工業革命以來,煤炭、石油、天然氣等化石燃料成為人類最主要的能源物質。化石燃料所產生的能源推動了社會生產力的快速發展,使得人類的科技水平、生活水平快速提升。然而,化石燃料的過度開采不僅破壞了大自然的生態平衡,同時釋放過量的CO導致溫室效應、海平面上升、海水酸化、局部地區氣候異常,給人類的生存帶來了極大的威脅和挑戰。
將大氣中的CO轉化為高熱值的可利用再生能源能有效解決能源短缺及CO含量過高的問題,近年來,電化學還原CO技術引起研究人員的廣泛關注,這種技術依靠可再生能源來獲取電能,將CO轉化為容易儲存的化學品以及工業原料,減輕了人類對傳統化石燃料的依賴;并且促進了CO的回收,遏制了CO的排放,能有效緩解過量CO排放導致的溫室效應問題。
電催化還原CO反應是通過電化學的方式將CO還原為化學品及燃料(如CO、甲酸、甲烷、乙烯、乙醇、丙醇等)的過程,一般由工作電極、對電極以及參比電極三電極體系構成;還包括離子交換膜,用于分離陰陽兩極室,避免電催化還原CO產物再氧化。依據體系電解液,電催化還原CO可以分為水溶液體系、有機電解液體系、離子液體體系三類。但由于有機電解液對環境的危害較大以及離子液體的價格昂貴,從而電催化還原CO的研究重心大多數都放在水溶液體系。在此體系中,通常選用廉價的無機鹽溶液(NaHCO、KHCO及NaSO等)作為電解質,能夠提供活化的質子,打破C==O鍵,免除了H原料。
1 電催化還原CO2基本原理
近年來,電催化還原CO快速發展,越發成熟,已被認為是一種有很大發展前途以及符合可持續發展理念的CO轉化技術。圖1 為電催化還原CO雙腔反應器,陽極(對電極)和陰極(工作電極)分別位于兩腔室中,陽極發生析氧反應,陰極為CO還原反應;兩腔室用質子交換膜(隔膜)分離,為了防止陽極產生的氧氣進入陰極腔室內氧化CO還原產物以及陰極CO還原產物進入陽極腔室內。普通陽極還原系統需要外界的電能來促進電子在外電路的傳輸,為了減少甚至消除外界施加的電能,一般通過可再生能源轉化為電催化還原CO系統所需的電能;光陽極和生物陽極是改進后的陽極,光陽極利用光能轉化為電能,生物陽極通過捕獲廢棄物種的化學能轉化為電能。
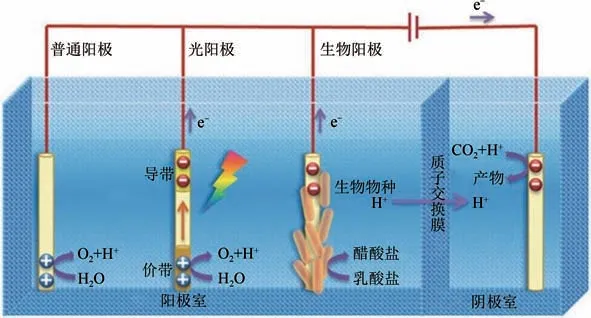
圖1 電催化還原CO2示意圖[8]
電催化還原CO是一個復雜的過程,通常發生涉及2~18 個不同電子轉移的反應,根據電子轉移數的不同,生成的產物一般有CO、CHOH、HCHO、CH、HCOOH、CH、CHOH、CHCOOH、丙醇等。表1為不同還原產物對應的化學方程式及其還原電位[相對于標準氫電極(SHE),pH=0、101.325kPa、25℃的水溶液]。此外,析氫反應作為兩電子反應過程,是電催化還原CO的競爭反應。

表1 不同還原產物的化學方程式及標準電勢
2 電催化還原CO2反應途徑
電催化還原CO的第一步是激活CO分子。因為負的氧化還原電位和穩定的分子結構,使CO分子得到一個電子變成活化的二氧化碳自由基中間體非常困難。但引入催化劑后,通過表面原子的相互作用,催化劑對CO分子通過化學鍵化學吸附,使得活化CO得到CO反應中間體相對容易很多。催化劑表面吸附的CO存在3 種可能的配位方式:①如圖2(a)所示,CO分子的氧原子有孤立電子對,可以與表面路易斯酸中心相互作用;②如圖2(b)所示,CO分子的碳原子可以從路易斯堿中心獲得電子,形成類似碳酸鹽的物質;③如圖2(c)所示,CO分子的碳原子和氧原子同時作為電子受體和給體,形成混合配位。CO分子吸附中間體不再具有線性對稱性,隨著CO分子彎曲,未占有電子能級最低軌道(LUMO)能級降低,從而CO分子吸附中間體具有較低的接受電子的勢壘。第二步是電子或質子遷移,打破碳氧鍵和形成碳氫鍵。最后一步是產物重排,從電極表面脫附,擴散到電解液。
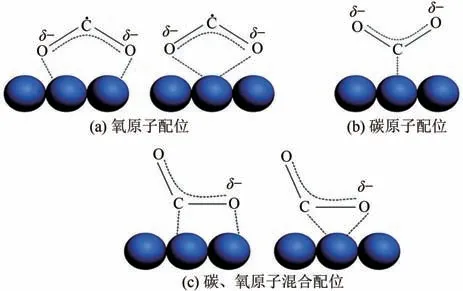
圖2 催化劑吸附CO2可能存在的結構形式[13]
3 電催化還原CO2不同產物形成途徑及活性催化劑
研究者們從實驗和理論兩方面探索了電催化還原CO生成C、C、C產物的形成途徑,大多數是基于Tafel 斜率或密度泛函理論DFT 計算結果的假設。電催化還原CO是一個比較復雜的過程,目前對于電催化還原CO的催化劑仍是一個挑戰,還沒有開發出具有足夠催化活性和穩定性的催化劑,距離大規模產業化仍然有比較大的差距。電催化還原CO的催化材料可以分為金屬材料和非金屬材料兩類,其中金屬催化劑最為普遍,尤其是過渡金屬催化劑。
3.1 C1產物
C產物一般是二、四、六電子轉移的反應,相對來說較為簡單,反應機理研究也相對成熟,報道出來的反應路徑機理被廣泛認可。Jens 教授課題組通過DFT 計算對電催化還原CO進行較為完整的研究,提出了C產物形成過程機理,并且結合原位表征手段,檢測催化劑原子結構變化、吸附分子反應成催化中間體變化等,明確了催化反應機理。
3.1.1 甲酸鹽和甲酸
電催化還原CO生成甲酸是通過關鍵中間體COOH 加氫得到,目前得到關鍵中間體COOH的線路中,圖3(a)、(b)最為成熟并且被廣泛認可。第一種形成路徑是一種中間體通過一個氧原子或兩個氧原子與金屬結合形成,如圖3(a)、(b)頂部路線所示。圖3(a)先是催化劑與氫離子作用得到吸附態的氫,隨后CO分子插入金屬-氫鍵與吸附態的氫反應形成中間體COOH。圖3(b)頂部路線是先得到吸附態的CO,隨后與溶液中的氫離子質子化形成中間體COOH。另一種途徑是通過二氧化碳自由基與臨近的質子通過碳與金屬結合形成關鍵中間體,如圖3(b)中部路線所示。研究表明,電解液中若存在HCO,則能夠提高甲酸鹽的產率,圖3(b)底部路線,是通過HCO提供的質子,與碳、金屬結合形成的關鍵中間體作用,得到甲酸鹽產物。圖3(c)是Bocarsly 等提出的一種表面結合碳酸鹽的路線,CO分子與表面羥基之間反應,形成表面結合的碳酸鹽中間體,隨后經過兩個電子和一個質子的轉移形成產物。CO分子較為穩定,在生成甲酸過程中,活化CO分子,形成關鍵中間體COOH是整個反應的速控步驟。
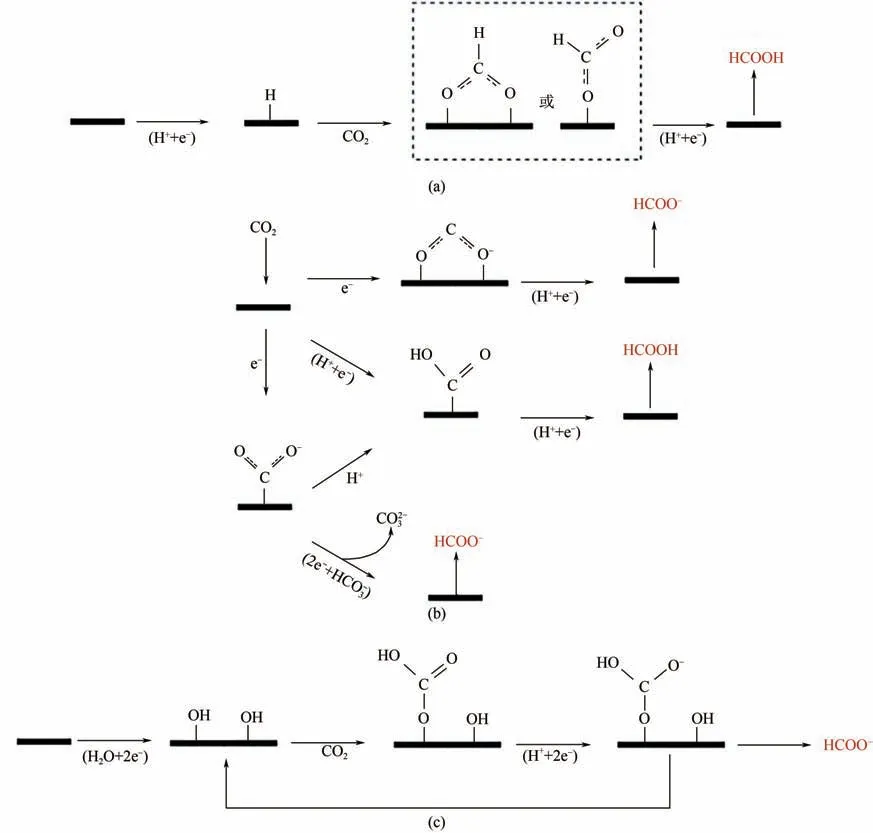
圖3 電催化還原CO2生成甲酸鹽和甲酸示意圖[18]
在反應過程中對關鍵中間體COOH 有較好選擇性的金屬催化劑有鉛(Pb)、錫(Sn)、銦(In)、鉍(Bi)等,產物以甲酸和甲酸鹽為主。在電催化還原CO所有產物中,甲酸產物最為簡單,反應機理相對成熟,被廣泛研究報道。譚勇文課題組利用電化學脫合金的方法制備了高導電性的Bi 金屬核和非晶態Sn 摻雜BiO殼的新型Sn 摻雜的Bi/BiO納米線催化劑,原位拉曼光譜和理論計算表明,Sn的引入能夠穩定生成甲酸的關鍵中間體COOH,抑制競爭性H和CO的產生,在寬的電位范圍內,都能表現出對甲酸產物很好的選擇性。朱起龍課題組首次組裝了一種自支撐的大尺寸三維多孔鉍系導電網絡,Bi-ene具有豐富的邊緣位缺陷,在寬的外加電位下表現出對甲酸鹽的高電流密度及穩定性,電流密度高達560mA/cm,連續運行500h 以上,活性無明顯衰減,并且原位紅外及理論計算進一步揭示了粗糙平面邊緣和平面內孔隙邊緣的豐富缺陷有利于COOH中間體的穩定。李彥光課題組利用簡單的溶液方法制備了表面碎片化的氧化鉍雙壁納米管,表面豐富的缺陷鉍位點,對生成甲酸有優異的活性、選擇性和穩定性,最顯著的是,在-0.61V下,其電流密度可達約288mA/cm,并結合DFT 計算,豐富的缺陷鉍位點能夠穩定COOH中間體,從而表現出優異的活性和選擇性。另外Li等利用溶劑熱法制備了尺寸均勻的PdBi 合金納米晶催化劑,PdBi 納米晶催化劑對中間體COOH有很強的吸附作用,對甲酸鹽的選擇性超過了90%,對CO 副反應起到很好的抑制作用。張華民團隊通過表面修飾法制備了Bi修飾Zn的Bi-Zn雙金屬納米結構催化劑,由于金屬-金屬雙功能界面和晶界提供了高密度的活性中心,在-0.8V時,甲酸的法拉第效率達到94%,且通過線性掃描伏安(LSV)測試得出,具有較高的催化活性。夏寶玉團隊對In作了研究,提出了一種氨基功能化的銦有機骨架,固定化的氨基增強了CO的吸收和活化,穩定了活性中間體,在-1.1V外加電壓下,電流密度為108mA/cm,有效地將CO還原為甲酸。
3.1.2 一氧化碳
電催化還原CO生成CO 是一個雙電子轉移過程,如圖4 所示,先是CO分子與質子反應活化得到COOH,隨后質子可攻擊COOH 的氧原子(OH)生成吸附的CO,進而脫附生成CO。Monteiro等通過還原產物CO 證明電解質中的正電荷粒子是穩定關鍵中間體的主要因素,對電解液中金屬離子的影響作用的微觀原理下了定論,陽離子能與吸附物配位,配位物與CO的靜電作用更能穩定CO,促進CO分子活化和催化劑表面電子轉移。生成CO反應過程中,除活化CO分子是一個限制反應的重要因素外,COOH 相較于CO 具有更弱的吸附能力,且受到尺度關系的限制,COOH 中間體加氫形成CO中間體的過程也是一個重要速控步驟。
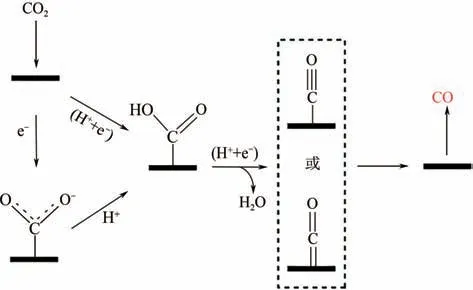
圖4 電催化還原CO2生成CO示意圖[18]
反應過程中,催化劑能夠選擇性生成COOH,并且對CO 吸附穩定性較弱,此類催化劑中有金(Au)、銀(Ag)、鋅(Zn)等金屬催化劑,有利于產物CO 的生成。Au 位于CO 演化動力火山圖頂部附近,在CO轉化為CO 方面具有較高的固有活性,被廣泛研究。Sun 等利用種子介導生長的方法合成了超薄的2nm Au 納米線,-0.35V 電位下,法拉第效率達到94%,并且保持6h,活性無任何變化,并且DFT 計算表明,Au 納米線的優異催化性能歸因于其活性邊緣位點的高質量密度和CO 在這些位點上的弱結合,這些超薄金納米材料是目前報道的最有效的電化學還原CO為CO 的納米催化劑。楊軍團隊利用兩步驟還原法精確合成了緊密堆積結構的金納米團簇,通過透射電鏡表征,團簇大小為1.5nm,通過電化學工作站測試的LSV 曲線及法拉第效率曲線,得到合成的納米團簇具有較高的活性,且在-0.57V 電位下,法拉第效率高達96%,高選擇性生成CO。Gao 等制備了Au-CeO結構,原位掃描隧道顯微鏡和同步輻射光譜研究表明,Au-CeO界面在增強CO吸附和活化方面起主導作用,DFT 計算表明,Au-CeO界面是CO活化和還原為CO的活性位點,Au和CeO之間的協同作用促進了關鍵羧基中間體(COOH)的穩定性,從而促進了進一步反應生成CO。除Au 外,Ag 也是被廣泛探索的將CO轉化為CO 的高本征選擇性電化學金屬催化劑,鹽酸溶液兩步脫合金法制備的納米多孔銀具有大的表面積和彎曲的內表面,在-0.6V 電位下,對CO 選擇性高達92%。除上述金屬催化劑外,非金屬催化劑也有報道,吳長征團隊報道了氮摻雜的石墨烯基催化劑,形成的孤立過渡金屬類環烷基是主要的活性位點,有利于CO 的生成。Kumar 等報道了利用聚苯胺碳化合成碳納米纖維,能夠將CO還原為CO,其優異的性能歸功于納米纖維的納米結構和關鍵中間體與碳納米纖維表面的高結合能,這一發現可能導致新一代的無金屬和非貴金屬催化劑,其效率遠高于現有的貴金屬催化劑。
3.1.3 甲醛、甲醇和甲烷
吸附態的CO 是生成甲醛、甲醇和甲烷的中間體,具體的途徑如圖5 所示。圖5(a)在得到吸附態的CO 后,先進行加氫反應得到CHO,再一步加氫,得到CHO(脫附生成甲醛),三次加氫得到吸附態的甲氧基(OCH),最后一步加氫還原脫附生成甲醇或甲烷產物。圖5(b)是另外一種生成CH的可能途徑,因為從動力學來看,C—O 鍵的解離和形成C—H 鍵生成CH的能量勢壘遠高于生成甲醇的能量勢壘,該步驟先經過兩步加氫還原反應從CO 得到吸附態的碳,再一步一步還原為CH、CH、CH,最后還原生成為CH。在該反應過程中,CO 加氫形成CHO 具有最高的結合能變,是反應的速率控制步驟,原因在于CO比CHO中間體在電極表面具有更強的結合能力。除DFT 計算及原位表征外,生成甲醇、甲烷產物,同位素標記也是確定反應中間體及反應機理的重要手段。
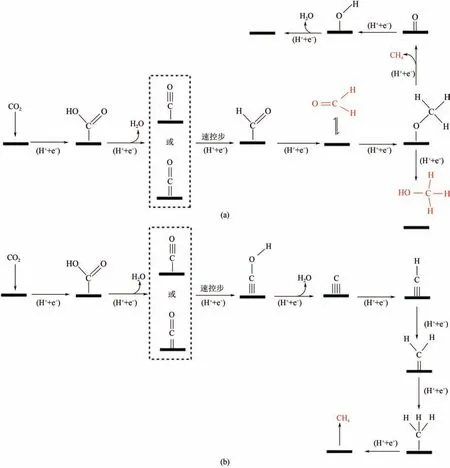
圖5 電催化還原CO2生成甲醛、甲醇和甲烷示意圖[18]
生成這些產物的催化劑對CO 有較強的吸附能力,在得到吸附態的CO 時,不容易脫去生成的CO,而是進一步加氫還原,得到甲醛、甲醇或甲烷。研究表明,Cu 在催化反應過程中可以吸附和轉化中間體CO,在不同的條件下可以產生多種不同的電催化還原CO產物,產物以甲烷為主。除金屬銅外,金屬氧化物CuO 和金屬Mo是良好的催化生成甲醇催化劑。Manthiram 等發現,在玻璃碳載體上分散良好的Cu 微球的甲烷化電流密度是高純銅箔電極的4倍,形成CH的平均法拉第效率為80%。黃昱團隊用電偶取代法合成了具有豐富Cu-Ag 界面的雙金屬Cu-Ag 納米線,首先合成Cu 納米線,然后通過從Cu 到Ag 的電流置換來實現Cu-Ag的原位合成,構建了具有豐富Cu-Ag界面的納米線,原子級的Cu-Ag 界面是提高甲烷的選擇性和降低過電位的有效方法。Ye 等制備出Cu摻雜的MoSe催化劑,生成CH具有較低的過電位,表現出優異的性能,并且利用DFT 對結構優化和電子結構性質進行了計算,研究結果有助于全面理解CO還原和CH選擇性對電子性能的影響,為進一步在原子水平上設計CO還原反應催化劑提供了重要指導。韓布興課題組首次報道了在缺陷CuO上構建原子分散的Sn催化劑,電還原CO得到甲醇的法拉第效率可達88.6%,是目前報道的最大法拉第效率,電流密度達68mA/cm,是迄今為止報道的活性最大的生成甲醇催化劑,Sn原子位置、相鄰氧空位和CuO載體配合良好,導致CO吸附容量更大,界面電荷轉移電阻更低,并且實驗結果和DFT 計算表明,該催化劑通過降低COOH 解離生成CO 的能壘,有利于CO的活化,得到的關鍵中間體CO與Cu結合進一步還原,導致對甲醇的高選擇性。鄭南峰團隊設計了一個二維層次Pd/SnO結構,部分由SnO納米顆粒包覆的超薄Pd 納米片,可以實現多電子轉移,選擇性還原CO成CHOH,這種結構設計不僅增強了CO在SnO上的吸附,而且由于Pd-O-Sn界面的構建,減弱了CO在Pd上的結合強度,這對提高Pd 催化劑的電催化選擇性和穩定性至關重要。
3.2 C2產物
C產物的形成是一個復雜的過程,可能包含多種途徑,這些仍是科研工作者探討和爭論的焦點,通過理論計算、原位表征、同位素標記及氫原子追蹤等方法確定了最合理的反應中間體及機理,圖6、圖7是較為成熟和被大多數科研工作者認可的途徑。

圖6 電催化還原CO2生成乙醛、乙烯和乙醇示意圖[18]
3.2.1 乙醛、乙烯和乙醇
吸附態CO 的形成是進一步還原為C產物的起點,經過關鍵的C—C 耦合作用,生成C產物。如圖6(a),先是四電子還原脫水生成CH,隨后可能是兩個CH耦合;或者CO 插入,經過電子還原脫水生成乙烯,這也是生成乙醇的途徑。CO 插入CH,經過還原可以生成乙醛和OCHCH中間體,OCHCH中間體進一步還原生成乙醇。圖6(b)是另外一種途徑,是通過CO共二聚與電子轉移形成CO關鍵中間體,隨后質子化成CO-COH,隨后通過還原生成乙烯、乙醛和乙醇產物。
3.2.2 乙酸鹽和乙酸乙酸鹽或乙酸的形成歸因于被吸附的CO-對還原產物CH的親核攻擊,CH的形成是通過吸附態的CO 還原而來,如圖7 所示。CO 先加氫得到CHO,再加氫得到CHO,再加氫還原脫水得到CH,最后再與被吸附的CO-反應,生成乙酸鹽或乙酸。
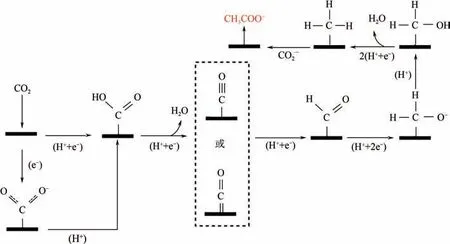
圖7 電催化還原CO2生成乙酸鹽和乙酸示意圖[18]
C產物過程不僅經過CO分子的活化,還有關鍵中間體CO 的形成,與C—C 耦合過程共同構成了該反應的速控步驟,C—C 耦合過程是生成C產物的關鍵。CO 是先決中間體,只有CO 的穩定生成,才會進行下一步的C—C耦合,最終生成乙烯、乙醇等產物。
由于C產物的復雜性,被報道的催化劑以Cu基為主,金屬Ag也有少量報道。Wu等制備了不同Ag殼層厚度的Cu@Ag核殼納米粒子,通過在Ag殼層上生成CO,進而在Cu 核上實現C—C 耦合,對C產物的法拉第效率高達67.6%,適當厚度的Ag層能有效地產生并提高Cu 核上的局部CO 濃度,有助于提高對C產物的選擇性,并且電化學測試和CO 吸附容量分析證實,Cu@Ag 催化劑在Cu/Ag界面上具有更強的CO 結合強度、更多的活性位點和更快的電荷轉移。Schuhmann等合成了以Cu基金屬有機框架材料(MOF)為自犧牲模板的新型納米結構CuOC電催化劑,利用聚四氟乙烯修飾納米電極,進行了局部活性測定,結果表明,該催化劑抑制了競爭性析氫反應,且對C產物的法拉第效率高達54%。Koper 等通過DFT 計算模擬結果發現Ag 有助于電催化反應生成乙醇。Yang 課題組提出了氯摻雜的多孔Cu 電催化劑,不僅C產物法拉第效率達到53.8%,而且很大程度提高了Cu基催化劑的壽命,在240h 都能表現出優異的催化穩定性,實驗結果表明,在電化學CO還原過程中,氯誘導活性位點豐富的結構是C+高選擇性的關鍵。Zhu 等通過電沉積銅絡合物原位還原制備了3D樹枝狀銅-氧化亞銅復合材料,這種結構充分暴露了活性位點,并且中間產物停留時間長,有助于多碳產物的生成,結果表明,C產物的法拉第效率高達80%,電流密度為11.5mA/cm,乙酸和乙醇的過電位僅分別為0.53V 和0.48V,具有優異的催化性能。Wang 等合理調整氮摻雜納米金剛石和銅納米顆粒的組裝,在外加電位僅為-0.5V 情況下,法拉第效率達到63%,且催化劑表現出前所未有的持續催化性能,達120h,電流穩定,活性僅下降19%,同時,DFT 計算表明,CO 在銅/納米金剛石界面的結合增強,抑制CO的解吸,促進C的生成。除金屬或金屬氧化物催化劑外,非金屬催化劑也被少量報道用于電催化還原CO生成C產物。
3.3 C3產物
迄今為止,電催化還原CO生成長碳鏈產物(如C產物)仍然是一個挑戰,生成C產物的僅有正丙醇、2-丙醇和丙酮。圖8為生成正丙醇的可能途徑,是通過吸附的C中間體與C中間體進行分子間的C—C耦合,再通過質子/電子轉移,還原生成正丙醇。
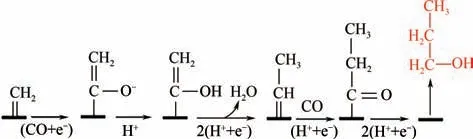
圖8 電催化還原CO2生成正丙醇示意圖[18]
目前,生成C產物的催化劑有石墨烯/氧化鋅/氧化亞銅,法拉第效率最大達到30%。Munir 等利用電沉積法制備了Cu-ZnO 催化劑,Cu-Zn 界面能快速促進C—C—C 耦合,高活性生成C產物;利用X 射線光電子能譜、拉曼光譜等進行元素分析,并通過活性測試提出了新的反應途徑,如圖9所示。CO分子得到3e和3H形成CHO,然后CHO 與C物質耦合形成吸附的C中間體,C中間體可通過左側途徑脫水加氫還原為正丙醇,也可通過右側途徑生成丙酮。生成2-丙醇的途徑,先是C中間體加氫使得吸附碳原子脫附,生成以氧原子為吸附中心的吸附C中間體,最后脫去邊緣羥基還原生成2-丙醇。
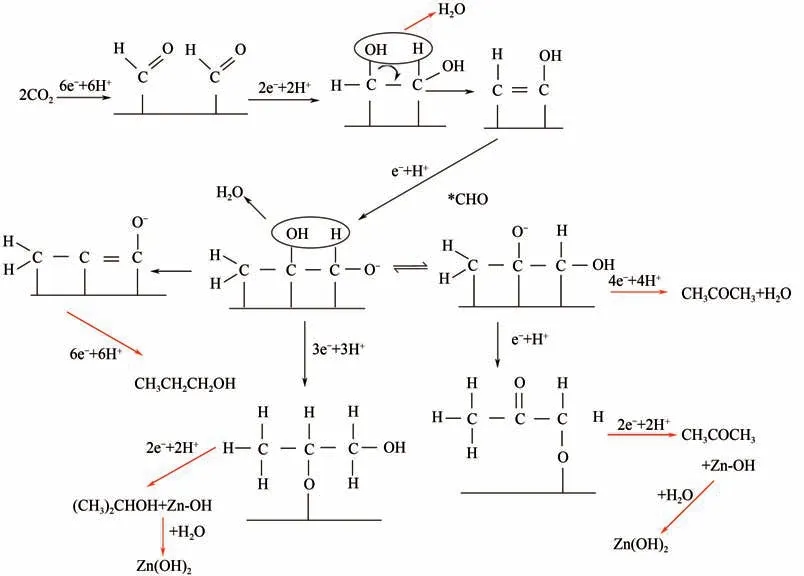
圖9 Cu-ZnO電極還原CO2生成正丙醇、丙酮和2-丙醇示意圖[55]
C產物更為復雜,反應經過多個決速步驟,需通過DFT 計算、原位表征(原位拉曼、原位紅外等)、同位素標記等手段,并結合電化學測試確定可能的反應中間體及機理;反應過程CO分子活化、CO的形成、C—C耦合、C中間體與C中間體的耦合等都是速控步驟,所以生成C產物的電流密度及法拉第效率較低,催化劑的活性和選擇性較低。
4 電催化還原CO2研究概況
目前電催化還原CO催化劑的穩定性和催化活性沒能得到有效解決,并且穩定性和催化活性較好的催化劑價格昂貴,很難大規模產業化應用。電催化還原CO有以下兩個較為突出的問題急需解決。
(1)生成C產物的低反應電流密度、析氫反應的競爭以及催化生成甲醇和甲烷選擇性問題。國內外對電催化還原CO生成甲酸及甲酸鹽產物催化劑研究較為成熟,制備出了活性及選擇性較高的錫(Sn)、鉍(Bi)等催化劑,在較低的外加電位下,活性較高,且對甲酸鹽的選擇性達到90%以上。對于C其他產物,都是通過吸附CO中間體轉化而來,通過圖10電催化活性金屬對CO及氫原子的吸附能,能夠得出金(Au)、銀(Ag)、鋅(Zn)對CO的吸附能力較弱,CO容易從催化劑表面脫附,產物以CO 為主,目前研究制備出的金(Au)、銀(Ag)等催化劑,對CO 的選擇性超90%,并且在CO 生成外加電壓下,電流密度較高。而Pb、Ti、In 等對CO 有較強的吸附能力,反應過程中會進一步加氫還原,得到甲醛、甲醇或甲烷。該過程相對復雜,目前被報道的催化劑選擇性較低,僅能達60%~80%。
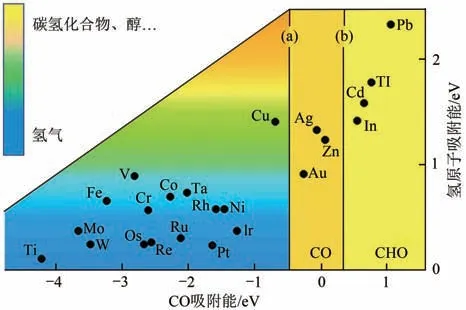
圖10 金屬電催化活性吸附能力圖[57]
綜合國內外報道的生成C產物催化劑,目前所能達到的法拉第效率如圖11 所示。在眾多還原產物中,CO、HCOOH 等C產物的研究較為成熟,在低過電位下的法拉第電流效率得到了很好的成果,但是反應的電流密度很低,很難達到預期效果。如增大電催化電流,在高的電流密度下,電極表面的CO會迅速消耗而得不到及時補充,這時競爭反應析氫反應的效率會顯著提高,使得產物大部分為氫氣,即降低析氫副反應效率,提高催化劑活性是研究重點。此外,對于甲醇和甲烷產物,形成過程相對復雜,反應過程法拉第效率較低,因此提高催化劑選擇性,即提高電催化生成甲醇和甲烷的法拉第效率也是研究難點。
(2)還原生成C及C產物的效率低及反應機理不明確問題。催化劑的選擇性以及催化劑促進C—C 耦合問題,都使得C及C產物形成效率處于較低的水平。Cu 催化劑是生成多碳產物的活性中心,因其反應過程的復雜性,C產物催化選擇性僅達50%,而C產物選擇性更是低于30%,如圖11所示。提高生成多碳產物催化劑的選擇性,增大法拉第效率是研究重點,探明反應機理,促進C—C耦合都是急需突破的問題。
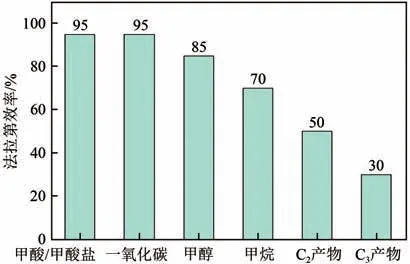
圖11 電催化還原CO2生成不同產物法拉第效率
5 電催化還原CO2催化劑改性方法
電催化還原CO生成C、C、C產物形成途徑中,除固有的單金屬催化劑外,為了提高催化劑催化活性、轉化率及選擇性,近幾年對于催化劑的改性被廣泛報道。不難發現,對于催化劑的改性主要有納米結構材料、負載型催化劑、雜原子摻雜、合金化、引入缺陷幾種方法。
反應物和反應中間體的吸附行為對催化劑的選擇性起著至關重要的作用,不同的改性方法會導致電子轉移途徑或速率發生改變,從而會導致產物的多樣性。目前,大多數吸附構型的調控都是通過特殊結構設計,優化催化劑的電子結構,降低反應中間體的形成勢壘來實現,上述的不同改性方法導致電子轉移途徑改變,從而導致產物的多樣性,所以合適的改性方法能夠避免副產物的過度產生,高選擇性地制備目標產物。
并且基于上述提到的電催化還原CO存在的問題,對于催化劑的改性來提高電催化還原CO的催化活性也是當前研究的重中之重。目前有兩種通用的方法來提高電催化劑的催化活性:①增加活性位點的數量。通過改進催化劑結構,暴露更多的活性位點;②提高每個活性位點的固有活性。為了達到催化劑最大催化活性,可以同時在這兩方面對催化劑改性,因為這兩種方法可以協同作用,不會相互排斥。制備納米結構或負載型催化劑材料可以大大增加活性位點的數量,而雜原子摻雜、合金化、缺陷的引入可以進一步提高每個活性位點的固有屬性。
5.1 納米結構
納米結構的催化劑具有更多的活性位點,如邊緣、臺階、角落等位置,此外具有大的活性比表面積,因此能表現出良好的催化活性。首先,不同尺寸的納米結構電催化劑具有不同的催化活性和產物選擇性,這被稱為催化粒徑效應。圖12 為不同銅納米尺寸還原過程的活性及選擇性,隨著粒徑減小,電流密度逐漸增大,即催化活性逐漸增加;粒徑較小,因其具有均勻的表面,有利于H、CO的脫附,選擇性生成H和CO,而隨著粒徑增加,有利于中間體加氫反應發生,即選擇性生成甲烷、乙烯等碳氫產物。
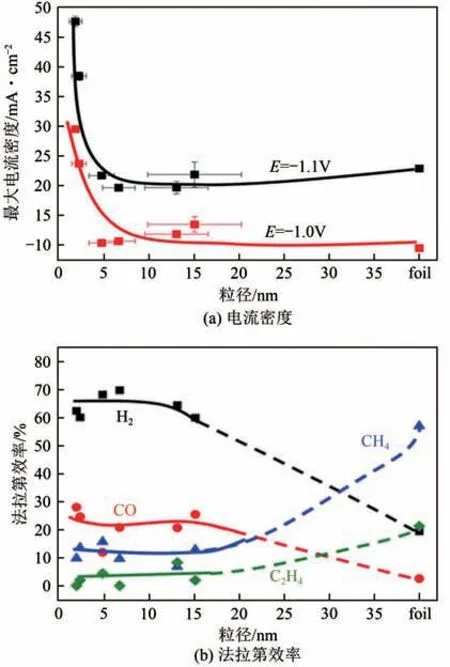
圖12 不同粒徑的銅納米粒子還原過程電流密度及法拉第效率[59]
究其原因,較小的Cu納米粒子直接影響角落、邊緣和表面的原子比例,增加了低配位的活性位點及電子傳輸速率,從而會引起催化劑的催化活性和選擇性,有利于CO 等的生成。大顆粒的結構具有粗糙的表面和孔/通道,這有利于電解過程中的電子傳遞和能量傳遞,還能使還原的中間體在孔/通道結構內進一步還原,生成碳氫產物,這種效應同樣在Au、Ag、Zn 等納米材料中存在。其次,對于電催化還原CO催化劑的改性可以通過制備不同形態的納米結構材料,這一方法是通過調整晶面、原子配位數對催化劑進行改性。同樣以Cu為例,探究Cu納米晶體選擇性-形貌的關系,3種晶形的銅被廣泛研究。立方銅主要暴露(100)晶面,傾向于將CO還原為乙烯;主要暴露(111)晶面的八面體銅,主要將CO還原為CO 和CH;而暴露(100)和(111)晶面的六方體銅有利于乙醇的生成,因為其存在大量的邊緣,這些邊緣可以調節吸附態氧原子的結合能,有利于將CO還原生成乙醇的過程。不同納米尺寸和晶型的晶體結構電子轉移途徑會發生改變,從而影響關鍵中間體的吸附,如小于5nm 的Cu 有利于CO 生成,大于25nm 的Cu 納米粒子更有利于生成甲烷、乙烯等產物,表現出不同的活性和選擇性。
5.2 負載型催化
催化劑的載體對于催化劑性能影響也至關重要,載體一般起到增加材料的比表面積、導電性和穩定性以及防止材料的聚集、保持材料性能的作用。目前被廣泛研究的載體有碳納米管、SiO、AlO、分子篩、TiO等,這些載體不僅能夠防止催化劑在催化過程中聚集,而且與催化劑存在化學偶聯的作用,能夠改變催化劑表面電子結構,影響電子轉移途徑,使催化劑的催化活性增強,并且提高了催化選擇性。以TiO載體為例,Ma 等制備了Ag 負載在TiO載體上的負載型催化劑,具體過程如圖13所示。
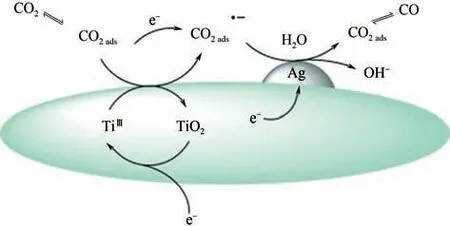
圖13 Ag/TiO2催化劑上CO2還原為CO途徑示意圖[65]
CO首先被吸附在TiO上,在較低的電位下,吸附的CO從電極上獲得一個電子(TiO氧化還原電子)形成活化的CO中間體,隨后CO從電極獲得一個電子(Ag 顆粒或載體TiO電子)生成Ag 吸附的CO,最終脫附生成目標產物CO;TiO是Ag納米顆粒的優越載體,不僅有助于Ag 納米粒子的分散,而且還穩定了還原性中間體,并作為氧化還原電子載體促進CO還原。Cu負載在NC薄層,NC層不會導致Cu 的電子結構變化,但是能夠選擇性的通過N-CO相互作用對CO進行富集和活化,提高Cu的催化活性。Ye等以MoSe為載體制備出Cu摻雜的MoSe催化劑,生成CH具有較低的過電位,表現出優異的性能,MoSe具有很好的活化CO作用,這是由于MoSe容易向CO轉移電荷造成的。負載型催化劑已成為一種普遍改性催化劑的方法,除被廣泛報道的載體外,新型載體SiO-TiO、MgO-SiO也逐漸進入研究者們的視野中,Cu 負載的這兩種新型載體都有很好的催化效果。負載型催化劑除增加材料的比表面積、穩定性等外,最主要的作用可能會提供電子,用于活化CO或者參與反應,促進中間體的生成和脫附,有利于催化劑的催化活性和提高選擇性。
5.3 雜原子摻雜
雜原子摻雜能夠改性催化劑的表面電子結構,或者摻雜的原子對某一關鍵中間體選擇性較好,從而提高電催化還原CO的催化活性。雜原子摻雜有金屬原子摻雜和非金屬原子摻雜兩種摻雜方式。實驗和理論計算表明,金屬原子摻雜的催化劑能降低反應中間體的形成勢壘,是因為其特定的電子結構和低配位。金屬原子摻雜主要是用于制備非均相單原子催化劑,并以碳基為載體,該結構是通過孤立分布的金屬與碳原子配位,降低反應中間體的形成勢壘,促進電子轉移,提高電催化還原CO的催化活性。除碳基載體外,譚勇文課題組制備的高導電性的Bi 金屬核和非晶態Sn 摻雜BiO殼的新型Sn 摻雜的Bi/BiO納米線催化劑,促進電子轉移,活性較高,并且表現出對甲酸產物很好的選擇性。非金屬原子摻雜也已被廣泛研究,B、N、F、P、S等非金屬原子已被成功的摻雜在石墨烯、碳納米管等碳納米材料中,該結構能夠誘導缺陷形成,因為碳原子和摻雜原子之間的電負性差異,破壞了原始的網絡結構。缺陷能夠提供更多的活性位點,并且會改變電子轉移途徑,進一步提高了催化活性。Xu等在MoS單分子層中引入Se原子導致Mo原子周圍的電子結構改變,使CO 產物的電導率和選擇性大大提高,遠遠優于MoS單分子層。雜原子摻雜會對催化劑的電子結構產生影響,提高活性位點電子密度,促進電子傳輸速率,并且降低中間體的形成勢壘,極大提高了催化劑活性和選擇性。
5.4 合金化
合金材料因為不僅能提供反應中間體的多個結合活性位點,而且有隨機可變的組分,所以合金材料是電催化還原CO極具吸引力的材料,圖14為單金屬和不同合金的催化選擇性結果。
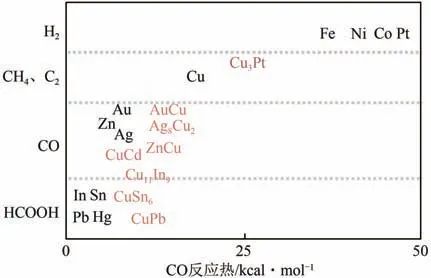
圖14 金屬和合金催化選擇產物[71]
與單獨的元素相比,合金材料能夠顯示出出色的活性和選擇性,可以通過調控不同元素的合金,利用金屬的不同催化作用及金屬界面的電子轉移途徑,制備出高活性及高選擇性的合金催化劑。Lim 等制備了不同Ag 殼層厚度的Cu@Ag 核殼納米粒子,利用Cu 和Ag 對不同中間體的吸附作用,通過在Ag殼層上生成CO,進而在Cu核上實現C—C耦合,提高了C產物的法拉第效率。合金的形成可能改變了與d中心理論相關的常規電子結構以及金屬活性位點的幾何和整體效應,有助于打破這些關鍵中間體的固有關系,從而改變選擇性。He 等研究了In與第二種金屬(Fe、Co、Ni、Cu、Zn等)的二元合金。因為In與H或CO的結合較弱,而第二種金屬與CO 結合能力較強,因此可以改變兩種金屬的比例來選擇性地生成產物。與單一金屬相比,銦合金不僅抑制了副反應氫氣產物的生成,而且提高了對CO 的選擇性,展現出良好的催化活性。合金化也會引起應變效應,改變金屬的價帶結構,從而改善催化劑的催化活性和選擇性。例如,在Cu表面原子中合金化少量Ag引起的壓縮應變效應極大地影響了Cu(100)的選擇性,更傾向于產生多碳氧化合物。合金化的金屬界面通常具有高的電子密度,并參與反應形成關鍵的吸附中間體,高活性和選擇性地制備目標產物。
5.5 引入缺陷
催化劑引入缺陷可以調節表面電子結構,并且缺陷部位通常被認為是電催化反應的活性位點,從而改善催化劑電催化性能。電催化還原CO催化劑引入缺陷的方法通常有浸出、等離子體處理、雜原子摻雜和電化學還原等。例如,通過從Au-Fe合金表面浸出Fe 原子,引入了許多缺陷,能降低關鍵中間體COOH 的形成勢壘,有效地增強了催化劑的活性和穩定性。等離子體處理可以增加催化劑表面粗糙度并在處理后的基體上產生缺陷,即使在室溫下也能迅速改變表面的化學狀態。例如,經過等離子體處理的Ag 箔和Cu 箔表現出比原始的Ag 箔和Cu 箔更好的電催化還原CO活性。周勇團隊通過簡單的探針超聲剝離相應的大塊晶體,然后在HO刻蝕下,引入了空位缺陷,提高了CO吸附和C—C 耦合能力,結果表明,催化劑表面粗糙度對催化劑的催化性能起部分決定作用,而銅的存在則是降低起始電位和提高乙烯選擇性的關鍵。缺陷的引入使得相鄰原子的化學狀態發生改變,電子密度受到影響,并且缺陷位點作為活性中心,電子傳輸相對容易,從而發生高活性催化反應。
6 結語
當今世界正面臨的能源短缺及過量CO導致的溫室效應問題,電催化還原CO生成容易儲存的化學品以及工業原料是一種有效方法。但目前電催化還原CO面臨幾個難題:①CO電催化反應產物生成的關鍵中間體及具體形成途徑仍需要不斷探索,技術面臨很大挑戰;②生成產物種類較多,得到具體某一產物較為困難,產物分離還需要大量配套工藝,對催化劑要求苛刻;③對于將來工業化來說,還有很長一段路要走,最關鍵的是催化劑還存在活性低、穩定性差、選擇性差、價格昂貴等問題。實現大規模產業化能有效解決能源及環境問題,其中最重要的是研究出活性好、價格低廉、制備便捷的催化劑。可通過增加活性位點數量和提高每個活性位點的固有活性兩方面改善催化劑。未來對于催化劑的研究可側重于以下幾點:①根據納米結構高比表面積的特點,著重研究各種納米結構催化劑;②探究出導電性好、比表面積大、與催化劑適配的支撐基板;③改變催化劑表面電子結構,影響電子轉移途徑,降低反應中間體的形成勢壘,高活性和選擇性制備目標產物。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
