CO2電催化還原產合成氣研究進展
2022-04-12 03:54:36華亞妮馮少廣黨欣悅郝文斌張保文高展
化工進展 2022年3期
關鍵詞:催化劑
華亞妮,馮少廣,黨欣悅,郝文斌,張保文,高展
(1 西安交通大學化學工程與技術學院,陜西 西安 710049;2 佛燃能源集團股份有限公司,廣東 佛山 528000)
2020年,我國提出的“碳達峰”“碳中和”目標對CO減排提出了更高的要求。電催化CO還原反應(COreduction reaction,CORR)是實現CO資源化利用,使其轉化為具有高附加值化學產品的一種有效方式,對減少溫室氣體排放具有重要意義,吸引了全球科學家們的重點關注。CO電催化還原反應在室溫常壓下進行,所需的電能可直接從太陽能、風能、地熱能、潮汐能等可再生能源中獲取,是一種將綠色能源轉化為化學能的高效儲能方式。CORR 是一個涉及多質子耦合多電子轉移的過程,通常包括2e、4e、6e、8e、10e、12e、14e等反應路徑,還原產物眾多,有C產物(CO、CH、HCOOH、CHOH)、C產物(CHCH、CHOH、CH、CHCOOH、CHCHO)以及C產物(CHCHO、CHOH)等。表1 總結了在標準大氣壓和25℃下部分CORR 產物及其在中性水溶液中相對標準氫電極(standard hydrogen electrode,SHE)的平衡電勢。
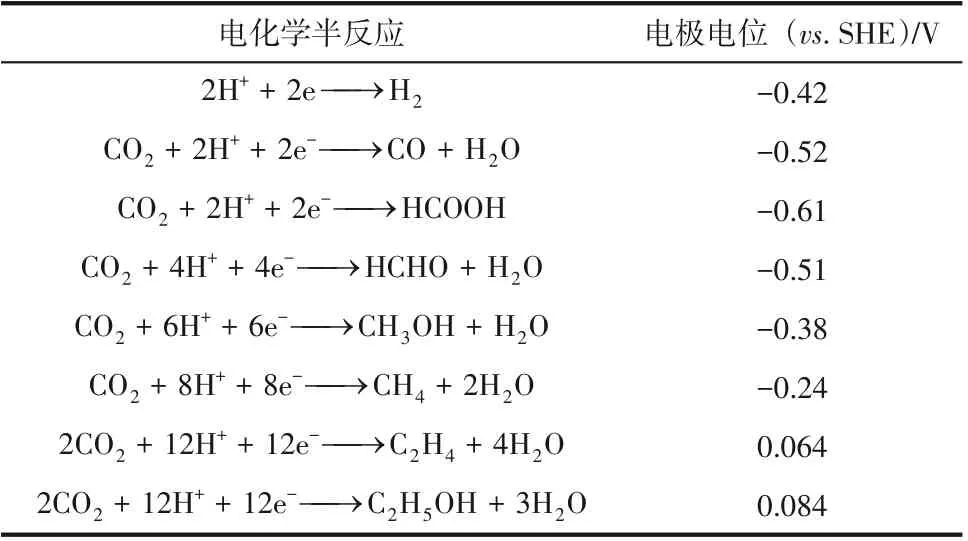
表1 電化學還原CO2半反應的電極電位(標準試驗條件)[10]
1 CO2 電催化還原產合成氣面臨的挑戰
CO電催化還原時,當以水溶液為電解質,由于陰極電解水發生析氫反應(hydrogen evolution reaction,HER),而HER 具有與CO電催化還原反應相當的熱力學電勢,因此在陰極CORR 與HER屬于競爭關系。當調控陰極CORR和HER反應時,則可以選擇性地生成CO和H的混合氣,即合成氣產物,它是石油化工行業重要的合成原料,可用于費托合成或生產甲醇等。傳統制備合成氣的方法包括煤氣化和天然氣重整等,需在高溫、高壓等極端條件下進行,消耗不可再生能源,與綠色化學的理念不符。利用CO和HO 作為原料,在水溶液中電催化還原CO,是可持續制備合成氣的理想方法,無需抑制析氫反應,也沒有液態產物分離困難等問題,成為當下的研究熱點。
盡管CO電催化還原制備合成氣在能源和環境方面具有獨特的優勢,但由于CO分子是一種熱力學穩定的線性分子,鍵長短、鍵能高、化學性質相對惰性,因此采用電化學方式還原CO產合成氣時,存在CO活化能高、反應過電勢高、選擇性差、催化活性低等問題。另外,由于不同的化工過程,所需的CO 和H組成比例不同,所以在CO電催化還原反應過程中調控合成氣組成比例,對下游合成氣的工業化應用極為重要。但目前仍然很難保證在高電流密度的同時實現寬范圍調控合成氣組成比例。因此,開發經濟、穩定、高效的電催化劑,通過催化劑結構設計,降低反應過電勢,提高催化活性,實現寬范圍精準調控合成氣組成比例,是推動電催化CO還原產合成氣技術應用的關鍵所在。
另外,水溶液中電還原CO產合成氣反應發生在氣、液、固三相界面處,涉及CORR 及HER 兩個反應,反應過程包含:①電解液中反應物CO向電極表面擴散;②CO和HO 被催化劑活性中心吸附;③電子和質子通過催化劑傳遞到催化劑表面的同時CO發生加氫加電子反應,產生COOH、CO、HO、H 等中間產物;④中間產物進一步轉化為CO、H等最終產物;⑤產物從催化劑表面解析等(如圖1所示)。因此,催化劑表面電子傳遞、加氫反應過程以及氣、液相傳質等都是CO電催化還原產合成氣反應的關鍵。根據Sabatier 原理,中間態物質與催化劑表面結合(化學吸附)既不太強也不太弱時,才會具有最佳的催化活性。因此,通過調節催化劑固有電子結構,獲得其表面對COOH、CO及H等中間產物最佳的化學吸附特性,調控CO 和H組成比例,可實現高效的CO電化學還原產合成氣反應催化活性。由此可見,CO電催化還原制合成氣反應的催化劑應當滿足以下要求:①對關鍵中間體COOH 具有適宜的吸附強度;②適宜的CO 吸附能;③CO 還原能力較弱;④原料豐富,價格低廉;⑤效率高,能夠同時生成CO和H。
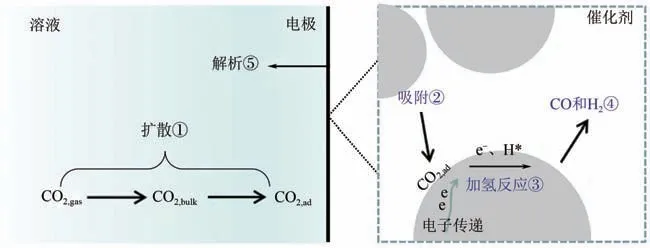
圖1 電極表面電催化反應過程
基于此,本文綜述了電催化還原CO產合成氣反應的催化劑研究進展以及用于電催化反應的電化學池特征。進一步地,總結了提高電催化還原CO產合成氣效率的策略,包括陽極反應耦合增強反應效率、設計雙活性位點電催化劑協同催化CORR、催化劑多級形貌調控增強反應傳質過程等,為電催化還原CO產合成氣技術工業化應用提供理論支持。
2 CO2電催化還原產合成氣催化劑
根據以上分析,由于標準條件下生成H和CO的電極電勢相似,在CO-HO 體系中,CO 和H都是電解產物。然而,催化劑的性質(如元素組成、結構、表面形貌、顆粒大小)和操作條件(如電壓、溫度、壓力、電解質的pH 等)對調節合成氣比例、提高合成氣生成效率起著重要的作用。幾十年來,開發高效、廉價、適宜的CO電催化還原產合成氣反應催化劑已然成為研究熱點。通過調節催化劑的大小、結構和組成,能夠提高催化劑的催化性能。在本節中,將討論5種不同類型的用于水溶液中CO電催化還原產合成氣的催化劑特征。
2.1 金屬催化劑
2.1.1 貴金屬催化劑
電催化CO還原產CO的催化劑中,由于Au和Ag 等貴金屬與CO 的結合能適中,不能進一步將CO還原為其他碳氫化合物,所以表現出較好的CO選擇性和催化活性。Hansen等構建了一個基于密度泛函理論計算的模型,通過催化劑對反應中間體CO和COOH的吸附能來描述CO電還原成CO的催化活性趨勢。由圖2(a)可以看出,Au 和Ag 具有較強的COOH 吸附能,同時具有較弱的CO 吸附能,從而表現出較強的CO電催化活性。水溶液中Au 催化CO生成合成氣的選擇性、活性以及合成氣組成比例等都與Au 納米顆粒的尺寸大小密切相關。Zhu 等研究了粒徑分別為4nm、6nm、8nm、10nm 的Au 納米顆粒電催化還原CO產CO 性能,結果表明,粒徑為8nm 時,Au 催化CO還原生成CO的選擇性最好[圖2(b)],且當粒徑分別為4nm、6nm、8nm、10nm 時,CO 和H比例分別為6∶4、7∶3、9∶1和3∶1。密度泛函理論計算結果表明,邊緣位點能夠催化生成CO,而拐角處的位點則是HER 反應的活性位點。邊緣與拐角位點的比例及活性位點數量均與Au粒徑大小息息相關。當Au納米顆粒粒徑大小不同時,其表面能和配位數不同,因此生成CO 和H的比例也不同,且當Au 納米顆粒粒徑越小、活性位點數越多時[圖2(c)],催化活性越強。
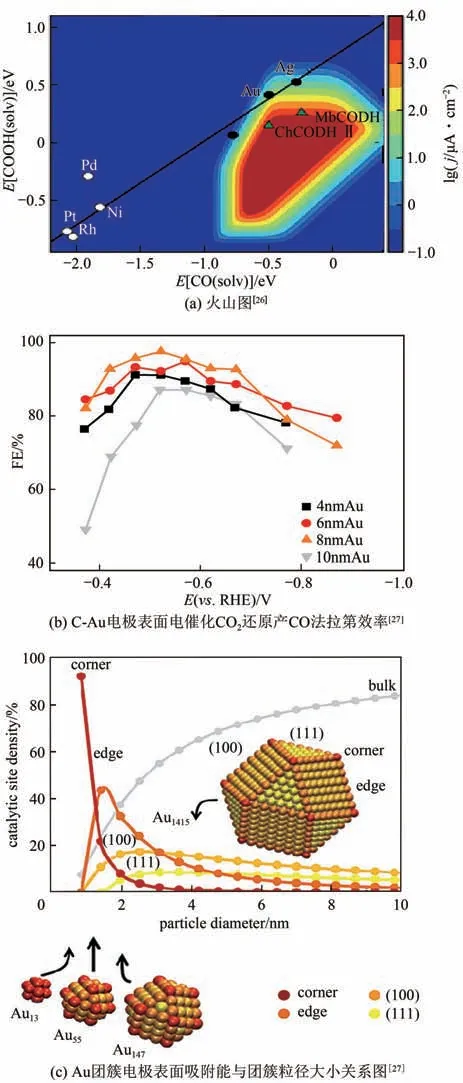
圖2 金納米顆粒催化活性
對于Au電極來說,其電催化CO還原生成合成氣的選擇性和活性,除與粒徑大小相關外,還受晶界(grain boundaries,GB)參數的影響。Feng 等采用氣相沉積法制備了Au/CNTs催化劑,其表面晶界密度受煅燒溫度影響。研究結果表明,Au/CNTs催化劑表面晶界密度與其催化CO還原產CO 選擇性呈線性相關,因此可以通過改變催化劑表面晶界密度調節合成氣產物組成比例。另外,合成氣組成比例(CO∶H)也可以通過改變Au納米線長度來調節。例如,當Au納米線長度分別為15nm、100nm、500nm,在電解電位(.RHE)為-0.4V 時,相應的CO∶H比例分別為1∶3、3∶1和9∶1。相比于Au 電極,Ag 電極因其具有較強的CO還原活性,價格較Au電極便宜,研究也較為廣泛。Liu等研究了三角形貌的Ag納米顆粒,其電催化CO還原,表現出高的CO選擇性和催化活性,CO的法拉第效率高達96.8%,原因在于三角形貌的Ag 納米顆粒具有更多的Ag(100)晶面,并且具有適宜的邊緣和轉角位點比例,合成氣的組成比例可以通過Ag 納米顆粒的形貌來調節(圖3)。

圖3 銀納米顆粒制備過程及催化機理[30]
近年來,關于CO電還原催化劑結構設計已經從抑制HER 反應到調控CORR 和HER 反應活性產合成氣,調節合成氣組成比例。Sheng 等利用商用Pd/C納米顆粒在水溶液中電還原CO生成組成比例可調的合成氣,當電解電位(.RHE)在-0.5~-0.6V時,得到CO/H比例在0.5~1之間,滿足工業上費托合成的需求。并且他們結合原位同步輻射和原位XRD 等,證實了Pd 在反應過程中與H 結合轉化成PdH的過程,PdH的形成能夠極大降低催化劑對中間產物CO 和H 的結合能,從而調節CO 和H的生成。Liu 等合成了過渡金屬氮化物負載的鈀納米顆粒(Pd/TMN)用于電催化CO還原產合成氣,取得優于商業Pd/C 的催化活性。通過原位XRD 及理論計算等證實,過渡金屬氮化物作為載體材料能夠有利于Pd 到PdH 的相轉變,PdH 的形成能夠降低CO 吸附能,避免Pd 表面CO 中毒。根據研究結果,5%的Pd/TMN 催化活性相當于10%的商用Pd/C,以過渡金屬氮化物為載體,顯著增強了Pd的催化活性和選擇性。
2.1.2 非貴金屬催化劑
由于貴金屬資源稀缺、價格貴,不利于大規模應用。因此,近年來,關于非貴金屬催化劑電催化CO還原產合成氣的研究越來越多。Qin 等采用電化學沉積法在泡沫銅表面合成了Zn 納米催化劑[圖4(a)],用于電還原CO產合成氣,在電解電位(RHE)為-0.7~-1.3V、溫度40℃、KHCO濃度0.1mol/L 時,生成CO/H的比例為0.5~1.2,電流密度11mA/cm[電解電位(.RHE)為0.9V]。根據研究結果,Zn(101)晶面表現出催化CO還原產CO 活性,而Zn(002)晶面則更容易發生析氫反應,因此,可通過調節Zn(101)與Zn(002)晶面比例來調節合成氣組成比例。Jeon等研究結果表明Zn納米顆粒催化活性和選擇性受催化劑粒徑大小影響較為顯著,當Zn 納米顆粒粒徑為3~5nm 時,Zn 催化劑表現出高催化活性,CO 產物法拉第效率高達70%以上,小于3nm 時主要為析氫反應,而大于5nm時,其性能類似于塊體鋅電極特性[圖4(b)]。計算結果表明,納米顆粒粒徑越小,其表面氫的覆蓋度越高,越易生成氫氣,通過調控Zn 納米顆粒粒徑,即可實現合成氣組成比例的寬范圍調節。
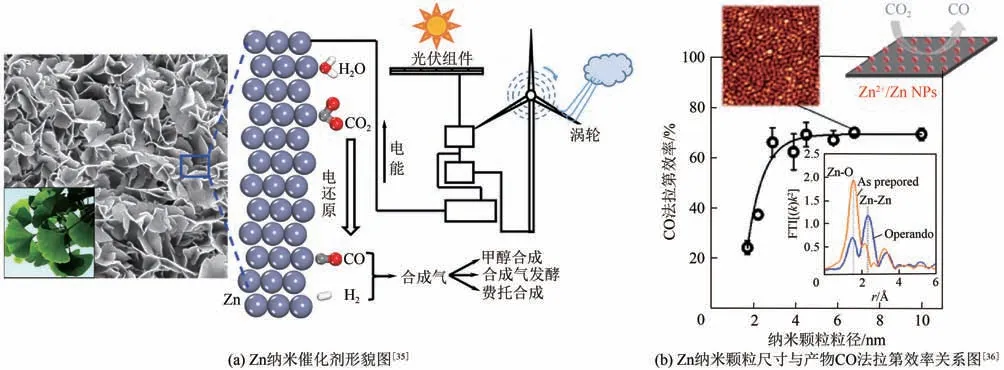
圖4 鋅納米顆粒電催化還原二氧化碳
雖然非貴金屬催化劑在電催化還原CO產合成氣領域有了一定的應用,但目前高效的催化劑主要還是貴金屬催化劑,非貴金屬催化劑的催化活性依然偏低,合成氣法拉第效率低,反應過電位高,電流密度低,且難以寬范圍調控合成氣組成比例。因此,仍需開發新型非貴金屬催化劑,提高催化活性,同時實現寬范圍調控合成氣組成比例。
2.1.3 合金催化劑
研究表明,多組分催化劑表現出與單組分不同的催化性能。由于不同組分之間的相互作用,物理和化學性質發生了改變,從而表現出不同的催化性能。合金可以調節催化劑電子結構及形貌特征,調節催化劑選擇性和催化活性,然而精確控制合金組成比例依然面臨很大挑戰。Fan 等通過在較大的負電流中進行電化學沉積,合成了SnPb合金催化劑。該合金催化劑具有多孔蜂窩狀主體結構和樹枝狀次級結構,CO電化學還原起始電壓比樹枝狀的Pb低80mV,在-1.16~-1.56V的電壓區間內生成甲酸法拉第效率接近100%。通過DFT 計算表明合金材料表面HCOO中間產物的吸附能較低,所以它的起始電壓較低。
通過合金催化劑還原CO和HO生成合成氣的研究報道相對較少。Watanabe 等在1991 年通過電鍍技術首次制備了雙金屬的銅合金(CuNi、CuSn、CuPb、CuZn、CuCd 及CuAg)催化劑,用于CO電還原產合成氣。研究結果發現,通過將低氫過電位的金屬位點引入高氫過電位的金屬表面(或反之),使反應物CO和H被吸附在相鄰的金屬位點上,從而降低反應過電位,調節CO和H組成比例。另外,過渡金屬(Fe、Co、Ni 等)與Au 的合金催化劑同樣能夠催化CO還原產合成氣,并且調節CO和H比例。Ross等合成了Fe基、Co基及Ni 基修飾的Au 電極電催化CO還原產合成氣,CO和H比例可以通過改變過渡金屬與Au的組成比例來調節。DFT 計算結果表明,Au 表面更有利于吸附COOH 中間產物,而過渡金屬元素3d 軌道更有利于發生HER 反應。該合金催化劑既能在更寬的電勢范圍調節合成氣組成比例,又能顯著增大反應的電流密度。
除了上述的二元合金外,三元合金也表現出較強的CO電催化能力,并且能夠調節CO 和H組成比例。He 等制備了硫硒鎘(CdSSe)合金納米棒催化劑,電流密度能夠在較寬的電勢范圍內都保持在10mA/cm以上。當CdSSe合金納米棒催化劑中不含Se時,CdS納米棒電催化CO還原,在電位(.RHE)為-1.2V時CO產物的法拉第效率最高達81%,電流密度為27.1mA/cm。隨著CdSSe合金納米棒中Se 含量逐漸增加,H的法拉第效率逐漸增大。Xu等合成了MoSeS合金催化劑,同步輻射結果表明,縮短的S—Mo 鍵和拉長的Mo—Se 鍵都有利于調節Mo 原子的d 帶電子結構。能量計算表明,Mo 原子周圍的偏心電荷不僅有利于COOH中間體的穩定,而且有利于CO 脫附。MoSeS 合金單分子層在電解電位(.RHE)為-1.15V時CO產率最高,法拉第效率達到45.2%,遠高于MoS單分子層(16.6%)和MoSe單分子層(30.5%)。研究結果揭示了三元過渡金屬TMDs合金中部分離域的電荷在原子水平上增強了催化劑電催化性能,為CO電還原催化劑開辟了新的研究領域。
2.2 金屬配合物催化劑
近年來,金屬配合物催化劑被認為是一類最具競爭力的電化學還原CO產合成氣反應催化劑,原因有:①金屬配合物催化劑(分子催化劑)對于一個既定反應過程表現出極好的活性、穩定性以及選擇性;②分子催化劑具有清晰的結構,能夠人為地設計和優化,達到更好的性能;③將分子催化劑負載在各種載體上可以用于制備單分散異相催化劑。非貴金屬配合物催化劑表現出優異的電催化CO和HO 還原制合成氣的能力,早在1980 年,Fisher等制備Ni、Co配合物催化劑用來在水溶液中電化學還原CO產合成氣,采用Co 配合物作催化劑時,CO 和H總的法拉第效率可以達到94%。Wang 等利用Ni(Ⅱ)三齒配合物均相催化劑[圖5(a)、(b)]電還原CO和HO,當電解液(DMF/HO)中含有5%的HO時,生成H的量可以忽略,主要產物為CO,當調節電解液中HO 的含量以及改變電解電位時,可以調控產物中CO 和H的組成比例,轉化頻數達1.9×10,催化劑可持續穩定反應一天。催化反應機理如圖5(c)所示。
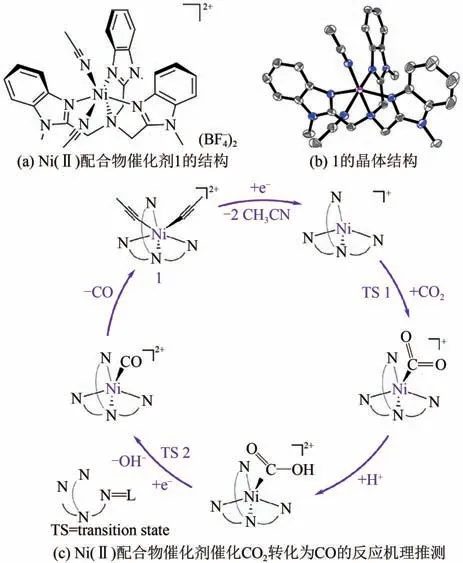
圖5 鎳配合物電催化還原二氧化碳[46]
貴金屬配合物催化劑同樣具有較高的電催化CO還原產合成氣催化活性。Wang等巧妙設計了雙功能的Ru 配合物催化劑[圖6(a)],陰極室中電催化還原CO和HO生成合成氣產物,同時陽極室中苯甲醇被氧化為苯甲醛[圖6(b)]。如圖6(c)所示,Ru(0)配合物作為反應中間物,不僅能與CO反應生成CO,還能與水反應生成H。CO 和H的組成比例可以通過調節電解液pH 以及電解電位來調控。
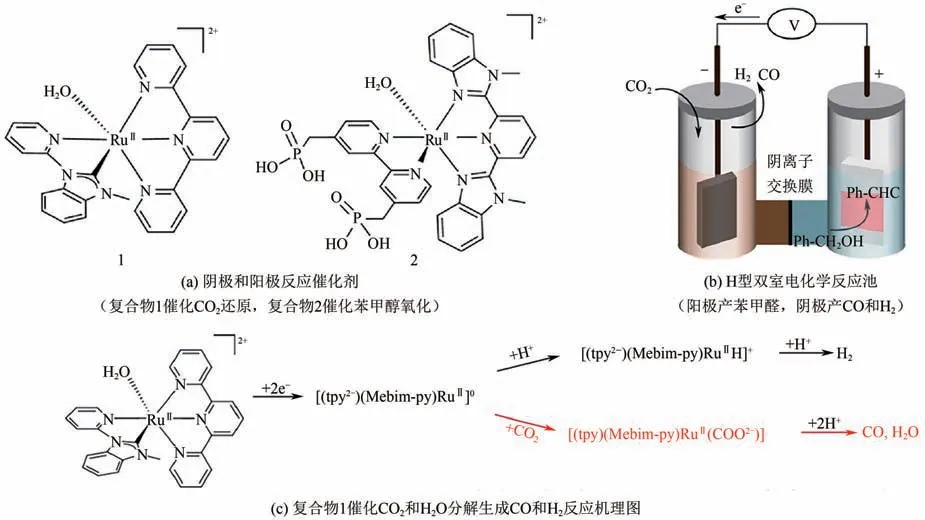
圖6 釕配合物催化劑電化學還原CO2產合成氣[47]
2.3 金屬氧化物和硫化物催化劑
電子從電子給體轉移至CO被認為是CO活化的關鍵步驟,因此通過調節催化劑表面電子結構來改善其催化活性是一種重要途徑。近年來,通過在非貴金屬催化劑中構建陰離子空位的方式來提高催化活性和調節合成氣組成比例受到了廣泛的關注。Qin等制備了硫化鎘修飾的碳納米管復合材料,其電催化CO還原生成CO 法拉第效率高達95%,通過電子順磁共振(EPR)及原位差分電化學質譜法等證實了在CORR 反應過程中,催化劑CdS-CNT 表面原位生成了硫空位。隨著硫空位數量增加,其電催化CORR 催化活性增強,電荷轉移阻抗顯著下降。結合原位紅外光譜及DFT 理論計算結果可知,硫空位能夠改變催化劑表面電子密度,降低中間產物COOH到CO轉變的能壘。因此通過改變硫空位的數量可調節CO和H的比例。同樣地,氧化物中的氧空位也能夠調節CO∶H比例。Geng等在富電子的催化劑表面通過引入氧空位來有效促進CO的活化。采用富含氧空位的ZnO納米片電催化CO還原產CO,電流密度高達-16.1mA/cm,CO 法拉第效率83%。理論分析表明,氧空位的引入增加了ZnO 的電荷密度,增強了CO的結合能,從而使得CO活化增強。
在以硫化物和氧化物作為催化劑電還原CO方面已有大量報道。Asadi 等證實二硫化鉬(MoS)具有較強的CO催化活性,MoS邊緣Mo原子因其金屬特性以及較高的d帶電子密度,使得催化劑具有較高的催化活性。通過調節電位(.RHE)-0.2~-0.8V,使得CO 和H的比例在1~99 之間得到有效調節。Rahman Daiyan 等利用火焰噴涂熱解法制備了含有晶體缺陷的ZnO納米材料,在連續流反應器中電催化還原CO產合成氣,當槽壓為2.6V 時,合成氣比值為1,電流密度可達40mA/cm。
2.4 金屬單原子催化劑
近年來,氮摻雜碳負載過渡金屬催化劑(MN-C,M=Fe、Co、Ni、Cu、Zn、Mn等)因價格低廉,具有CO電催化還原活性,成為最具潛力的貴金屬催化劑替代品之一。根據負載型金屬納米催化劑的尺寸效應,金屬尺寸的減小不僅能夠增加金屬位點的不飽和配位環境,增強金屬本征活性,而且又能增強金屬與載體及吸附物之間的相互作用,提高催化活性。當金屬尺寸縮小至極限即單原子時,其催化活性呈指數增長,因此,原子級分散的M-N-C催化劑成為近年來CO電催化劑的研究熱點之一。原子級分散的M-N-C 催化劑,作為一種單原子催化劑(single atom catalysts,SACs),相比于塊體及納米催化劑,具有原子級高度分散的活性位點、配位不飽和度高、均一的配位環境等特點,使其在電催化CO還原反應中呈現出高原子利用率、高催化活性及選擇性等優勢,表現在:①SACs表面均一的特性使得其對CO和H產物具有優異的選擇性;②催化劑表面原子利用率可達100%;③在合成氣形成過程中有利于檢測活性位點及反應路徑。
近幾年來,研究者們在過渡金屬M-N-C 單原子催化劑電催化CO還原產合成氣方面取得了較大的突破。通常過渡金屬納米顆粒具有較好的HER活性,因其資源豐富,廉價易得,成為貴金屬Pt催化劑最具潛力的替代品之一。研究發現,當過渡金屬元素(Fe、Co、Ni 等)以單原子形式均勻分散在載體上時,其具有極強的電催化CO和HO生成合成氣的活性。Yang 等通過熱解法制備了Ni 單原子催化劑,通過高角度環形暗場掃描透射電子顯微鏡(HAAD-STEM)表征了Ni原子尺寸只有0.2nm,Ni 單原子價態在0~2 之間。相比于氮摻雜石墨烯負載的金屬鎳納米顆粒,氮摻雜石墨烯負載的Ni單原子催化劑電催化CO還原過電位下降了200mV,當電解電位(.RHE)為-0.95V 時,CO和H的比例為9∶1,遠高于Ni 納米顆粒。Ni 單原子催化劑穩定性長達100h。Song 等利用Co-CN單原子催化劑電還原CO,當電解電位(.RHE)在-0.7~-1.0V 時,合成氣法拉第效率可達100%,CO/H的比例為0.5。
另外,金屬單原子價態也會影響其催化CO還原產合成氣性能及合成氣組成比例。Gu 等報道了單原子分散的Fe-N-C 催化活性遠高于單原子分散的Fe-N-C,研究結果表明,三價態的Fe 在N摻雜碳載體中能夠與吡咯態氮配位,在催化反應中一直保持Fe形式。當CO 部分電流密度為1.0mA/cm時,Fe-N-C 需要的電位(.RHE)為-0.525V,而Fe-N-C需要的電位(.RHE)僅為-0.285V。另外,當電解電位(. RHE)從-0.2V下降到-0.45V 時,Fe-N-C 電極能夠調節CO∶H的比例為(2∶1)~(18∶1),而Fe-N-C 電極調節的CO 和H比例僅為2~4。相比于Fe,Fe具有更強的CO吸附能力,而對CO 的吸附較弱,因此有利于CO生成。
由于單原子負載型催化劑中,金屬單原子與載體間存在強相互作用(strong metal-support interaction,SMSI),可使電子在載體和金屬之間發生傳遞,從而影響催化劑表面化學吸附性能,另外,通過調控載體的多孔結構,有利于改善物質傳輸性能。因此,作為單原子催化劑載體須具備以下幾個方面的功能:①具有能適合反應過程的形狀和大小;②有足夠的機械強度和抗拉強度,能夠經受反應過程中的機械沖擊;③有足夠大的比表面積,以均勻負載足夠多的活性中心;④合適的孔結構,以利于氣、液、固等多種形態的反應物和產物的傳遞過程;⑤穩定性好,以抵抗活性組分、反應物及產物等的化學侵蝕。
2.5 非金屬催化劑
快速發展的碳材料因其自然資源豐富、穩定性好以及環境友好,被認為是最具潛力的產合成氣非金屬電催化劑。另外,碳材料不僅具有較大的比表面積、超高的電導率,并且具有多孔結構,孔隙率高,從而有利于CO的吸附以及電解液的快速滲透。但是天然的碳材料具有較低的活化CO和HO分子的能力,而熱解的碳材料通常對于共還原CO和HO活性較低。然而,當摻雜了其他雜原子(N、S、B、O 等)時,會調節碳材料內部電子結構,從而表現出優異的產合成氣催化活性。Kumar 等首次報道了非金屬碳材料作為電催化劑同時還原CO和HO,通過靜電紡絲技術利用聚丙烯腈(PAN)制備了碳納米纖維(CNFs),采用CNFs 作為催化劑電催化CO,當電解電位(.SHE)為-0.573~-1.14V 時,產生合成氣產物,其中,吡啶氮和三維石墨烯結構在催化反應過程中起到了關鍵作用。Ji等采用等離子體技術處理的氮摻雜碳納米管陣列,作為電催化劑,通過電化學CO-HO 還原制備CO/H比例可控的合成氣產物。在不同的等離子體處理條件下,CO/H比例范圍可達0.55~3.03,適用于下游化工生產的原料氣標準。通過優化等離子體處理條件,CO 的法拉第效率最高可達75%,并且能夠維持穩定性長達10h。根據研究結果可知,碳納米管中摻雜的吡啶氮有利于使CO轉化為CO,而吡咯氮和sp2平面外的碳則有利于H的產生。因此,利用等離子體處理的方法能夠有效調節催化劑中各活性組分的比例,從而調控CO還原反應和HER 的速率,最終實現CO/H比例可控的合成氣產物。
通常認為在雜原子摻雜的碳材料中,雜原子為催化活性位點,但近期的研究報道指出,被雜原子活化的碳原子是電催化CO還原產合成氣的活性中心位。Xie 等研究報道了在以F 摻雜的碳電極電催化還原CO時,F 元素的摻雜能夠活化與其鄰近的碳原子,一方面有利于增強中間產物COOH 在電極表面的穩定性,另一方面能夠抑制HER過程,從而有利于CO到CO 的轉變過程。當電解電位(.RHE)在-0.5~-1.0V 時,純碳材料電還原CO時沒有活性,而對于F 元素摻雜的碳材料電還原CO時,可以調節CO 和H產物的比例,得到合成氣組成比例在(3∶14)~(9∶1)之間。
3 CO2 電還原產合成氣的電化學反應池
3.1 H型電解池
CO電還原反應通常在三電極體系中進行,即采用H 型電解池(圖7),中間采用離子交換膜將陰陽極室隔開以利于CO還原產物的收集檢測,同時避免產物的再氧化。陽極電解水生成氧氣,即發生氧的生成反應(oxygen evolution reaction,OER),陰極室CO接受電子和質子發生還原反應,同時,陰極電解水生成氫氣。但由于水溶液中CO溶解度低(0.033mol/L),在傳統的H型電解池中傳質受限,使得CO電還原產合成氣反應電流密度低,一般不超過100mA/cm,使該技術在實際應用中受限。
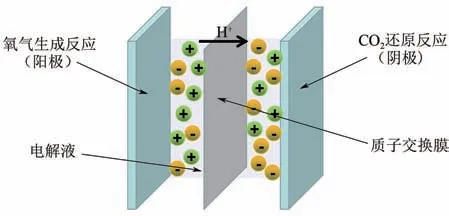
圖7 典型的H型CO2電還原系統
3.2 連續流電解池
通過在氣室和液室之間使用氣體擴散電極(gas diffusion electrode,GDE),可以緩解催化劑表面的氣液界面物質傳輸問題。Hara 等采用GDE組裝了Co、Ni、Rh、Pt、Pd、Cu及Ag電極,用于CO電還原產合成氣,CO氣體壓力小于50atm(1atm=101325Pa)。在Ag-GDE電極體系中,當CO壓力為20atm時,CO產物法拉第效率達86%,電流密度高達300mA/cm;當壓力大于30atm 時,電流密度高達3.05A/cm。而對于Pd-GDE 電極,電流密度從0~1000mA/cm,CO 和H比例在4~1 之間。Xiang 等采用原位電化學沉積的方法首次制備了GDE表面修飾的Cu-In電催化劑。這種核殼結構的Cu-In催化劑電還原CO時產物CO法拉第效率高達90%以上,當電解電位(.RHE)為-1.17V時,電流密度高達200mA/cm以上,使得2.0cm電極表面上CO 產率可達3.05mg/min(CO 流速為15mL/min)[圖8(a)、(b)]。
為解決H型電解池中,由于傳質受限而導致的電流密度偏小問題,研究者們在氣體擴散電極的基礎上開發了連續流反應器。Xie 等制備了Bi-Pd單原子合金(single atom alloy,SAA)納米枝晶(nanodendrites,ND)催化劑,其中Bi 原子單分散在Pd矩陣中,用于CO電還原產CO 反應中。在以BiPd-SAA ND為催化劑時,對比了H型電解池和連續流電解池,其CO 法拉第效率分別為90.5%和91.8%,過電位分別為290mV和200mV[圖8(c)~(e)],在連續流反應器中,電流密度高達173mA/cm,遠高于H型電解池(1.9mA/cm)。
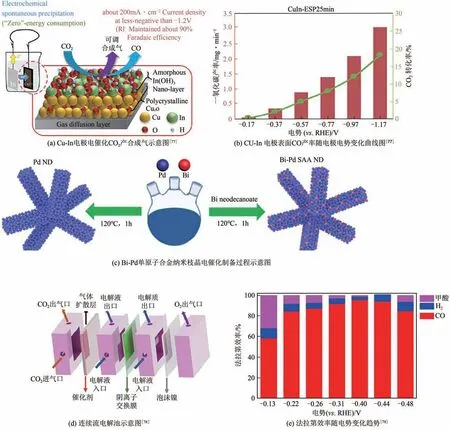
圖8 合金催化劑電還原CO2
3.3 固體氧化物電解池
固體氧化物電解池(solid oxide electrolysis cell,SOEC)可以將CO和水轉化為合成氣及烴類燃料等。電解池具有全固態和模塊化結構、能量效率高、成本低等優點,在CO轉化和可再生清潔電能存儲方面極具應用潛力(圖9)。鈣鈦礦型陶瓷陰極由于在氧化還原氣氛下結構穩定,且可有效抑制積炭反應,是近年來SOEC 領域的研究熱點。然而,鈣鈦礦型陶瓷陰極氧空位濃度低,CO吸附弱,CO活化和轉化困難,導致CO電催化還原性能較低。Zhou等制備了釩摻雜的鑭鍶鐵與釓摻雜的氧化鈰納米復合材料(LSFV/GDC),作為SOEC陰極應用于高溫CO電催化還原反應。實驗和理論計算結果表明,釩的摻雜可增加陰極氧空位濃度,提高了陰極CO高溫吸附活化能力和電催化還原性能。在800℃、1.6V 時,SOEC 電流密度可達0.62A/cm,比未摻雜時提高了51.2%,法拉第電流效率接近100%。該研究通過金屬元素摻雜來調控SOEC陰極材料氧空位濃度和CO吸附活化能力,為提高SOEC 陰極CO電催化還原性能提供了新思路。Gaudillere 等研究了Cu 基SOEC 電解池,用來電解CO和HO 產合成氣,反應溫度為600~700℃。由結果可知,SOEC 體系中,在中溫區電解CO和HO產合成氣是可行的。
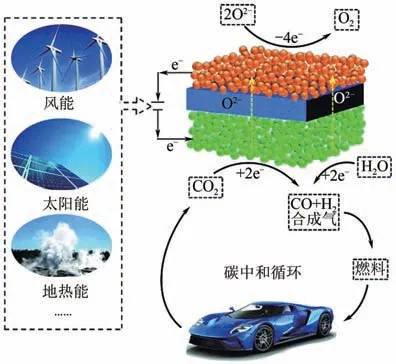
圖9 高溫CO2電解的碳循環過程[79]
SOEC 電解CO的成本在很大程度上取決于耗電量,電力成本占SOEC 電解CO總成本的80%以上。因此,開發高性能和穩定的SOEC,降低電力消耗,對提高其經濟競爭力至關重要。為此,SOEC 電還原CO技術未來的研究主要集中在以下幾個方面:①基于DFT 計算,利用原位表征技術監測電極反應的基本過程和SOEC的衰減過程,為進一步探索高活性電極材料提供指導;②通過調節雙鈣鈦礦氧化物、相結構以及引入活性界面,增強材料性能進而促進電化學反應;③通過將甲烷氧化偶聯、低碳烷烴氧化脫氫、甲烷芳構化或其他化學反應耦合到SOEC 陽極,實現SOEC 多功能化,不僅可以促進陰極CO電化學還原,而且能夠充分利用陽極產生的氧物種實現烷烴高效轉化;④降低SOEC系統內部阻抗,提高系統效率等。
3.4 膜反應器電解池
由于H型電解池在應用過程中存在槽壓高、液態產物有效濃度低等問題,因此,研究者們開發設計了基于質子交換膜燃料電池(proton exchange membrane fuel cell,PEMFC)的膜電極構型的電解池,其具有易于模塊化且可規模化實現CO電催化轉化的優勢,應用潛力巨大。采用陰離子交換膜電極組件(anion membrane electrode assembly,AMEA)可以使電流密度極大提高。Jiang 等研究了氮摻雜石墨烯負載Ni 單原子催化劑(Ni-NG)電化學還原CO產合成氣的催化活性。當采用膜電極組件時,電流密度可高達50mA/cm,Ni-NG 能夠連續反應8h。在實際應用中,通過增大氣體擴散電極(GDL)尺寸、增加催化劑載量以及增多膜組件數量等方式,能夠增強AMEA膜電極電化學池轉化效率。
在基于膜電極的電解池中,當使用陽離子交換膜(CEM)時,陽極必須在酸性介質中,需要使用貴金屬OER 催化劑,增加成本,且易使陰極發生析氫反應,當使用陰離子交換膜(AEM)時,通常采用碳酸氫鹽作為電解質,陰極產物易從陰極轉移至陽極。通過將CEM 和AEM 同時層壓在膜電極表面,制備成雙極膜(BPM),能夠顯著改善上述問題。基于BPM的電解槽可以通過將H和OH離子分別選擇性地傳輸到陰極和陽極來維持兩側的恒定pH,增強催化反應活性及電解池穩定性。Li 等研究了基于雙極膜組件的電化學反應池中,采用非貴金屬催化劑電還原CO產合成氣,電流密度可高達200mA/cm。在基于雙極膜組件的電解池中,能夠實現對質子的有效管理,從而能夠使系統穩定運行,并且在高電流密度下實現CO的電催化反應(圖10)。
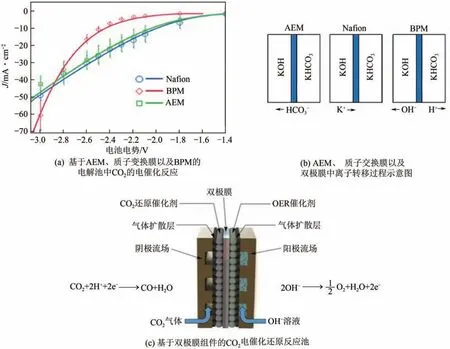
圖10 基于雙極膜電解池的CO2電還原技術[89]
4 產合成氣效率提升策略
4.1 陽極反應耦合
典型地,電化學CO還原產合成氣體系包含陰極的CORR、HER 和陽極的OER 過程。電化學還原CO過程中,電子從陰極電極表面轉移到溶液中的CO分子或CO溶劑化離子中。由于將電子提供給CORR 的陽極OER 反應動力學過程緩慢,根據Hess 定律,反應體系70%的能量消耗都是來自OER過程,因此以OER作為陽極反應會導致CO電催化體系較大的過電勢和較高的總能量消耗。由于多數的電催化CO還原產合成氣體系都只考慮了陰極半反應,而沒有考慮體系整體的電流效率、過電勢和能量效率。因此,在陽極室以熱力學上有利的反應(醇氧化、尿素氧化等)來代替陽極OER,能夠增強電解池整體的能量效率,降低槽壓。Verma 等通過共電解CO和丙三醇,采用丙三醇氧化反應代替OER時,可使電能消耗降低53%,生成產物乙二醇和乙醇。Li 等通過在Cu-In 電極上電還原CO產CO,電流密度為3.7mA/cm,CO法拉第效率大于70%;同時陽極鉑電極表面采用醇氧化代替OER 反應,可生成比氧氣更具有實際應用價值的產物,產率>75%[圖11(a)]。
特別地,使用合適的電解質和活性催化劑,在同一電化學反應器中同時從陽極和陰極產生相同的產物更具有應用前景。Wei 等以過硫酸銨為氧化劑,在氫氧化鈉溶液中一步氧化法制備了泡沫銅表面負載氧化銅納米片(CuONS/CF),并在陽極上電催化部分醇氧化(MOR)為甲酸;同時合成了碳布上生長的介孔二氧化錫(mSnO/CC),并將其作為電化學還原CO的高效陰極催化劑,生成甲酸。在陽極上用動力學上有利的部分MOR 取代OER,可以顯著降低陽極和電池的電位輸入。在甲醇電解液中使用CuONS/CF 作為陽極催化劑,電流密度達到10mV/cm時的電位比OER 降低250mV,法拉第效率達到95%[圖11(b)]。此外,mSnO/CC具有較高的催化活性,電催化CO還原產甲酸的法拉第效率可達81%。當陽極電極為CuONS/CF、陰極電極為mSnO/CC 時,所構建的電解槽僅需0.93V 的槽電壓即可達到電流密度10mA/cm,相比相同催化條件下,電池電壓降低500mV。
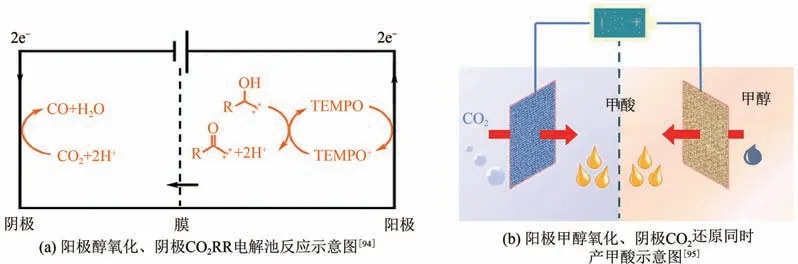
圖11 陽極反應耦合陰極CO2還原反應
4.2 雙活性位催化劑結構設計
電催化CO還原產合成氣反應過程中,催化劑表面同時發生著CORR 及HER 兩個反應過程,CORR 過程產生COOH、CO 等中間產物,HER 過程會形成H 等中間物質,根據Sabatier 原理及以上分析,中間態物質與催化劑表面結合(化學吸附)既不太強也不太弱時,才會具有最佳的催化活性。因此,通過調節催化劑固有電子結構,獲得其表面對COOH、CO、HO 及H 等中間產物最佳的化學吸附特性,調控CO和H組成比例,實現高效的CO電化學還原產合成氣反應催化活性。
通過雙活性中心協同調控催化劑表面對合成氣中間產物適宜的吸附能,對提高電還原CO產合成氣反應的催化活性具有重要意義。He 等利用葡萄糖、雙氰胺以及過渡金屬鹽溶液混合物作為前體,制得Co、Ni 雙金屬單原子催化劑,當電解電位為-0.5~-1.0V(RHE)時,CO/H比例為0.23~2.26,理論計算結果表明,Ni SAs 為產CO 活性中心,而Co SAs 是產H的活性中心。Song 等通過熱解ZnO@Zn/Co-ZIF 納米球前體,利用過量Zn 的揮發,抑制熱解過程中Co原子團聚,獲得了Co質量分數高達3.4%的Co-CN單原子催化劑,其Co原子表面電子結構有利于吸附CO分子,能夠拉長C=O雙鍵,活化CO分子,是CORR的活性中心,催化劑中吡啶氮和石墨氮等含氮官能團則是HER反應活性中心。在雙活性位催化劑中通過調控雙活性位點的比例來調節合成氣組成比例。
另外,根據文獻報道,過渡金屬單原子催化劑具有較高的催化CO還原生成CO活性,而Fe、Co、Ni 等金屬納米顆粒則具有較好的析氫活性。Jiang 等研究對比了氮摻雜石墨烯負載Ni 單原子催化劑(Ni-NG)與石墨烯負載Ni 納米顆粒(Ni-NPs/G)電催化還原CO活性,結果表明Ni 納米顆粒幾乎只有析氫活性,而Ni 單原子還原CO時,CO 選擇性高達97%。基于此,Daiyan 等構建了鈷單原子協同納米顆粒(Co@CoNC-900)的雙活性位型催化劑,通過Co 單原子與納米顆粒間相互作用,調控催化劑固有電子結構,從而得到組成比例可調的合成氣產物,實現了高效電催化CO還原。
為了構建過渡金屬雙活性位型催化劑,有研究者采用沸石咪唑酯骨架材料(zeolite imidazole frameworks,ZIFs)為前體/模板,利用ZIF 的孔道限域及配位鍵合等方式固定金屬原子。因ZIFs 具有豐富的氮源和可調控的孔道結構,其有機單體對金屬的錨定作用以及籠狀ZIFs 對內部金屬的封裝效果都有利于限制金屬原子在高溫炭化過程中的遷移與團聚,這對于形成高分散的金屬活性位點有極大的幫助,通過調控熱解過程實現金屬單原子(single atoms,SAs)協同納米顆粒(nanoparticles,NPs)雙活性位型催化劑的構筑。通過構建過渡金屬NPs/SAs 雙活性位型催化體系,協同調控CORR和HER 催化活性,從而獲得可調比例的合成氣產物。
4.3 催化劑多級形貌調控
針對CO電還原產合成氣反應過程中三相界面處的傳質問題,通過采取分級形貌結構調控,可獲得更高密度的、均勻分散的活性位點以及互相連通的導電框架,實現電催化劑表面傳質過程的強化。研究者多通過零維(0D)碳納米球、一維(1D)碳納米管和二維(2D)石墨烯等碳材料相互作用,巧妙構建多級結構框架作為碳載體,以增強反應催化效率。Sun等利用MOF媒介拓撲轉化策略在泡沫鎳(NF)上構建了多孔的三元CoFeNi LDHs 框架,其孔洞邊緣部分含有豐富的缺陷位及未配位的金屬活性中心[圖12(a)]。這種多級結構的形成增大了催化劑比表面積,獲得更多的活性位點。Yan等采用模板法在碳納米棒上原位生長了Ni納米顆粒和氮摻雜碳納米管,形成了Ni@N-CNT/NRs分枝狀的多級結構,具有豐富且分散均勻的活性位點、較大的比表面積、較小的電荷轉移和物質擴散阻力以及較強的機械穩定性[圖12(b)]。Pan 等采用氧化開壁的方法使多壁碳納米管部分開壁,形成緊密結合的碳納米管/納米帶復合結構,最終制備了Fe、N共摻雜的Fe-N/CNT@GNR框架[圖12(c)],因其較高的電化學比表面積、較強的物質轉移能力及豐富的多孔結構,使其具有高電催化CO還原活性,CO法拉第效率可達96%,電流密度為22.6mA/cm。
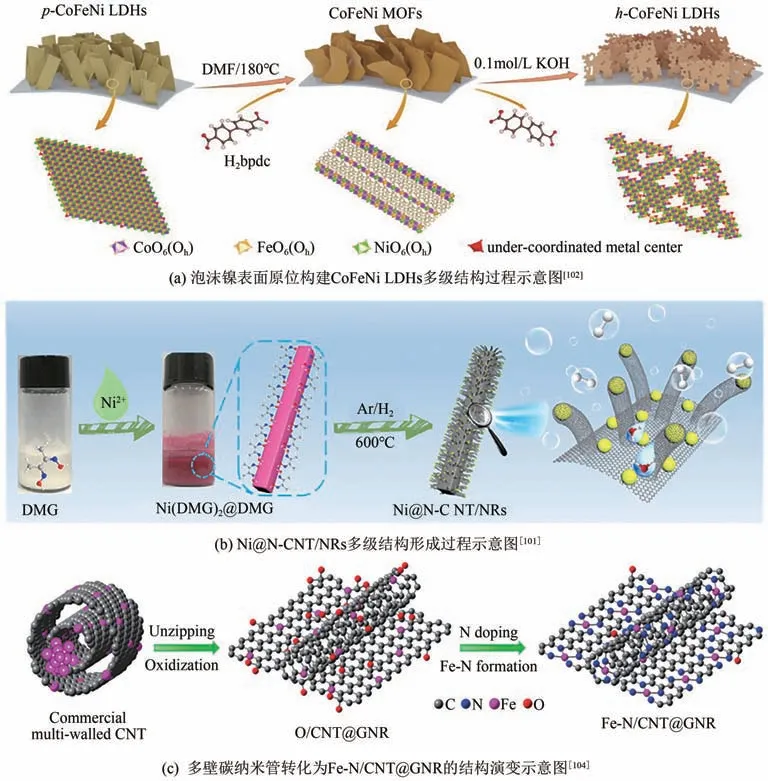
圖12 碳多級結構
碳納米管是一種中空管狀的一維(1D)碳結構材料,電子轉移能力強,石墨烯作為一種具有超大比表面積及超強導電能力的二維(2D)材料,也備受學者青睞,碳納米管和石墨烯作為單原子載體已有較多的研究報道。有研究者結合石墨烯和碳納米管的結構特性,構筑了類似三明治夾心的多級(1D/2D)碳結構負載過渡金屬電催化劑,具有以下幾方面的優勢:①可提高催化劑比表面積,增加活性位點密度;②碳納米管和石墨烯超強的導電性,可增強催化劑電子轉移能力,促進CO電還原反應;③三明治夾心的多級碳結構,有助于形成豐富的孔結構,強化氣、液傳質過程;④可避免多孔碳雜亂堆疊和導電性差的問題,同時碳納米管、石墨烯之間的界面結構對于金屬活性位的形成以及催化活性的提高也有重要意義;⑤有助于提高催化劑穩定性。
5 結語和展望
電化學還原二氧化碳(CORR)利用可再生能源,將CO資源化,生成高價值的燃料和化學品,是有助于實現全球可持續能源經濟的一項有前途的策略,但在實現低成本、大規模商用之前,仍有眾多問題值得關注。
第一,亟需開發新的方法來加快高活性催化劑的設計和篩選。新興的機器學習和高通量實驗方法的發展為催化劑預測篩選能力提供了解決途徑,有助于材料發展和CORR電催化劑的設計。
第二,現有理論基礎對于描述電化學界面電荷的遷移,以及在反應條件下評估反應動力學和反應能壘方面仍然存在挑戰。多尺度模擬有助于更好地了解真實反應中的界面過程,是增強實驗和理論研究相融合的有效手段。
第三,結合原位技術,包括原位衰減全反射表面增強紅外吸收光譜(ATR-SEIRAS)、原位表面增強拉曼散射、原位同步輻射、原位X射線光譜等技術深入探究反應中間產物、反應機理等。
第四,開發低成本、高性能催化劑,增大電流密度,增強催化活性及選擇性,提高催化劑穩定性。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
