LC-MS/MS檢測(cè)豆芽中的4種植物生長(zhǎng)調(diào)節(jié)劑
2022-04-12 14:42:20黃景怡王宗義黃丁偎董子琦張嘉彤
北京農(nóng)學(xué)院學(xué)報(bào) 2022年2期
黃景怡,郭 凱,王宗義 ,閆 珺,黃丁偎,董子琦,張嘉彤,魏 朦
(北京農(nóng)學(xué)院 食品科學(xué)與工程學(xué)院/食品質(zhì)量與安全北京實(shí)驗(yàn)室/農(nóng)產(chǎn)品有害微生物及農(nóng)殘安全檢測(cè)與控制北京市重點(diǎn)實(shí)驗(yàn)室,北京 102206 )
豆芽,俗稱豆芽,是人們?nèi)粘I钪匾氖巢闹弧=陙恚寡恐兄参锷L(zhǎng)調(diào)節(jié)劑(PGRs)殘留問題受到了人們的廣泛關(guān)注[1]。PGRs作為一類人工合成的激素農(nóng)藥,具有提高作物產(chǎn)量和改善產(chǎn)品品質(zhì)的作用[2],但過量的PGRs殘留,容易導(dǎo)致兒童早熟、女性生理改變、老年人骨質(zhì)疏松等,影響生殖和代謝,甚至有致癌、致畸的作用[3-5]。2014年,中國(guó)開始不受理PGRs在豆芽制發(fā)中使用的登記申請(qǐng)[6-7];2015年4月, 又明確規(guī)定6-芐基腺嘌呤(6-BAP)、4-氯苯氧乙酸(4-CPA)和赤霉素(GA)等不在豆芽生產(chǎn)可使用的范圍[8]。但近年來豆芽中PGRs仍有不同程度檢出的報(bào)道[9-12],因此,建立豆芽中典型PGRs的簡(jiǎn)便可靠測(cè)定方法, 對(duì)提高相關(guān)食品安全監(jiān)管效率具有實(shí)際意義。
目前,關(guān)于豆芽中PGRs的檢測(cè)方法主要有氣相色譜法(GC)、高效液相色譜法(HPLC)[13-14]和液相色譜-串聯(lián)質(zhì)譜法(LC-MS/MS)[10-11, 15]等;其中GC法需要衍生化處理,較為繁瑣,HPLC法通常靈敏度和選擇性不足,LC-MS/MS法是近年來報(bào)道較多的方法,具有高靈敏、高選擇的特點(diǎn),但容易受基質(zhì)效應(yīng)的影響,需要優(yōu)化合理的樣品前處理方法。本研究,在Sutcharitchan等[16]檢測(cè)三七中多種PGRs的基礎(chǔ)上,針對(duì)豆芽樣品進(jìn)行了適當(dāng)方法改進(jìn),建立了豆芽中4-CPA、6-BAP,GA和2,4-D等常被檢出PGRs的高效檢測(cè)方法。
1 材料與方法
1.1 材料與試劑
黃豆、綠豆、黃豆芽、綠豆芽(北京地區(qū)超市和農(nóng)貿(mào)市場(chǎng));50-mL塑料離心管(BD Biosciences公司)。
分析純無水硫酸鎂、氯化鈉、檸檬酸三鈉、檸檬酸二氫鈉(國(guó)藥集團(tuán)化學(xué)試劑有限公司);色譜純乙腈、甲醇、甲酸、乙酸銨(美國(guó)J.T.Baker公司); 4-CPA(濟(jì)南中天組培有限公司)、GA、6-BAP、2, 4-D(上海源葉生物有限公司)。
1.2 儀器與設(shè)備
Agilent6410B液相色譜-串聯(lián)質(zhì)譜聯(lián)用儀(美國(guó)Agilent Technologies公司);Centrifuge 5810 R高速冷凍離心機(jī)(德國(guó)Eppendorf公司);KQ-500 DE超聲波清洗器(昆山市超聲儀器有限公司);800 A型樣品均質(zhì)機(jī)(天津泰斯特儀器有限公司);MS 3基本型漩渦混勻器(艾卡(廣州)儀器設(shè)備有限公司);Heal Force純水機(jī)(默瑞(上海)生物科技有限公司)。
1.3 樣品前處理
將采集好的新鮮豆芽用榨汁機(jī)打成糊狀,準(zhǔn)確稱取10 g(精確到0.001 g)于50 mL離心管中,加入20 mL乙腈,渦旋混勻1 min,超聲10 min,冷卻至室溫后加入4.0 g MgSO4、1.0 g NaCl、1 g Na3Cit·2H2O和0.5 g Na2HCit·1.5 H2O,再次渦旋混勻、超聲10 min,冷卻至室溫后,9 500 r/min離心5 min,取部分上清液過0.22 μm的有機(jī)濾膜后,再取部分濾過液加等體積的去離子水混合后,裝入至進(jìn)樣瓶中待測(cè)。
1.4 標(biāo)準(zhǔn)溶液的配制
分別準(zhǔn)確稱取4-CPA、GA、6-BAP、2,4-D標(biāo)準(zhǔn)品各10 mg,分別于10 mL容量瓶中, 用甲醇定容, 得1.0 mg/mL各標(biāo)準(zhǔn)儲(chǔ)備液,于-18 ℃保存。
用甲醇稀釋標(biāo)準(zhǔn)儲(chǔ)備液,獲得濃度均為10 μg/mL(加標(biāo)回收試驗(yàn)用)和1 μg/mL混合標(biāo)準(zhǔn)中間工作液于4 ℃保存,使用時(shí)放置到室溫。
分別取5 mL空白黃豆芽樣品提取液于10 mL容量瓶中,再分別加入1 μg/mL的混合標(biāo)準(zhǔn)工作液各 2 000、1 500 、1 000、500、200和100 μL,用去離子水定容至刻度,得200、150、100、50、20和10 ng/mL的系列標(biāo)準(zhǔn)混合溶液。同樣方法配制綠豆芽樣品基質(zhì)系列標(biāo)準(zhǔn)混合液。
1.5 分析條件
色譜分離:色譜柱為Waters XEELECTTM HSS T3 (3.5 μm, 2.1 mm×150 mm);流動(dòng)相A為含0.1%甲酸水溶液;流動(dòng)相B為乙腈;梯度洗脫程序?yàn)?5%的B保持2.5 min,然后1.5 min 線性升至80%,保持7 min, 重新回到35%,平衡色譜柱7 min;進(jìn)樣體積20 μL; 柱溫40 ℃; 流量0.3 mL/min。
質(zhì)譜檢測(cè): ESI+電離源,霧化電壓15 psi,去溶劑氮?dú)鉁囟葹?00 ℃、流量10 L/min,碎裂電壓135 V;多重反應(yīng)監(jiān)測(cè)(MRM)模式質(zhì)譜檢測(cè),各化合物的前級(jí)離子質(zhì)荷比,定量產(chǎn)物離子質(zhì)荷比(碰撞能),定性產(chǎn)物離子質(zhì)荷比(碰撞能)分別為:6-BAP,m/z226、m/z91(20 eV)、m/z226(0 eV); 4-CPA,m/z185、m/z127(10 eV)、m/z129(10 eV);GA s,m/z345、m/z239(15 eV)、m/z143(15 eV);2,4-D,m/z219、m/z161(20 eV)、m/z125(20 eV)。
1.6 數(shù)據(jù)處理
使用MassHunter B.07.01數(shù)據(jù)處理軟件,對(duì)目標(biāo)物保留時(shí)間、母離子及兩個(gè)二級(jí)離子響應(yīng)比值與標(biāo)樣相應(yīng)參數(shù)進(jìn)行對(duì)照,進(jìn)行定性;建立基質(zhì)標(biāo)準(zhǔn)曲線,外標(biāo)法進(jìn)行定量,定量計(jì)算公式:w=c×V×n/m,其中w為試樣中待測(cè)物含量(mg/kg),c為最終樣品溶液中待測(cè)物的濃度(ng/mL),V為提取液體積(mL),n為稀釋倍數(shù),m為樣品質(zhì)量(g)。
2 結(jié)果與討論
2.1 色譜與質(zhì)譜條件的優(yōu)化
在不接色譜柱的條件下分別用濃度為1 000 ng/mL的標(biāo)準(zhǔn)溶液進(jìn)行全掃,找到每個(gè)目標(biāo)物的前級(jí)離子,然后在不同碰撞能條件下進(jìn)行碎片離子掃描,確定每個(gè)化合物的碎片離子。試驗(yàn)發(fā)現(xiàn),4-CPA和6-BAP僅能獲得1個(gè)相對(duì)豐度較大的碎片離子,因此,根據(jù)Cl元素有較大的27Cl同位素豐度的特點(diǎn),分別使用185和187做前級(jí)離子,而6-BAP則使用其前級(jí)離子在碰撞能為0 V時(shí)做為碎片離子之一,進(jìn)行MRM條件設(shè)置,獲得較好效果。
在優(yōu)化的MRM參數(shù)下,比較了0.1%甲酸-乙腈,0.1%乙酸-乙腈,0.1%甲酸-甲醇,0.1%乙酸-甲醇的分離效果和流動(dòng)相對(duì)檢測(cè)信號(hào)響應(yīng)的影響。試驗(yàn)發(fā)現(xiàn),0.1%甲酸-乙腈,效果最好,乙酸雖然使4-CPA等負(fù)離子模式檢測(cè)的目標(biāo)物響應(yīng)有所增加,但分離效果不佳,甲醇則洗脫時(shí)間較長(zhǎng),故選擇0.1%甲酸-乙腈進(jìn)行梯度洗脫。黃豆芽和綠豆芽樣品基質(zhì)對(duì)分離沒有影響,典型MRM色譜圖如圖1所示。試驗(yàn)為排除基質(zhì)效應(yīng)的影響,使用基質(zhì)匹配標(biāo)準(zhǔn)溶液建立標(biāo)準(zhǔn)曲線。
2.2 樣品前處理方法的改進(jìn)
Sutcharitchan等[16]使用水-乙腈提取,結(jié)合鹽析做用,進(jìn)行樣品前處理,檢測(cè)了中藥三七中39種植物生長(zhǎng)調(diào)節(jié)劑,樣品前處理方法較為簡(jiǎn)單實(shí)用。不同于中藥材,豆芽含有大量的水分,因此豆芽樣品可直接進(jìn)行乙腈提取,不需要加入水。試驗(yàn)發(fā)現(xiàn),樣品經(jīng)乙腈提取,硫酸鎂脫水后的提取液,直接進(jìn)樣分析,色譜峰展寬嚴(yán)重,色譜效果較差;將提取液用水稀釋到兩倍體積時(shí),色譜峰型變好(圖1)。固本試驗(yàn),采用樣品提取前不加水,而提取后,加水稀釋進(jìn)樣的樣品處理方式,獲得了理想的試驗(yàn)效果。

圖1 豆芽基質(zhì)中4種待測(cè)目標(biāo)物的MRM色譜圖Fig.1 The MRM chromatograms of 4 PGRs in soybean sprouts
2.3 線性、檢出限和定量限
結(jié)果(表1)顯示黃豆芽和綠豆芽基質(zhì)中驗(yàn)證濃度為10~200 ng/mL,標(biāo)準(zhǔn)曲線線性良好,R2>0.995。分別以3倍信噪比和10倍信噪比計(jì)算檢出限(LOD)和定量限(LOQ),結(jié)果見表3,LOD為0.07~5.62 μg/kg,LOQ為0.25~18.74 μg/kg。
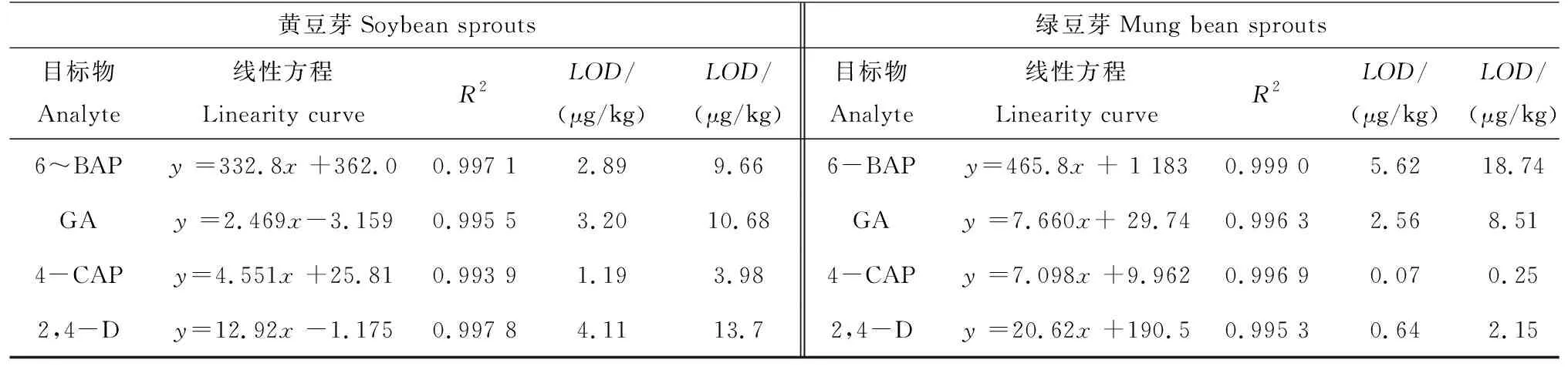
表1 線性、檢出限和定量限TableTab.1 Linearity, LOD and LOQ
2.4 加標(biāo)回收率與精密度
按1.3小節(jié)的樣品處理方法,稱樣后分別加入10 μg/mL的混合標(biāo)準(zhǔn)工作液20 μL、50 μL和150 μL,獲得20、50、150 μg/kg的樣本,每個(gè)水平6個(gè)重復(fù),渦旋混合30 s,然后按1.3小節(jié)后續(xù)處理方法處理,并檢測(cè),結(jié)果見表2。4種目標(biāo)物的回收率為73.81%~109.57%, 相對(duì)標(biāo)準(zhǔn)偏差RSD為3.71%~16.88%,符合化學(xué)分析方法驗(yàn)證確認(rèn)和內(nèi)部質(zhì)量控制實(shí)施指南要求[17]。
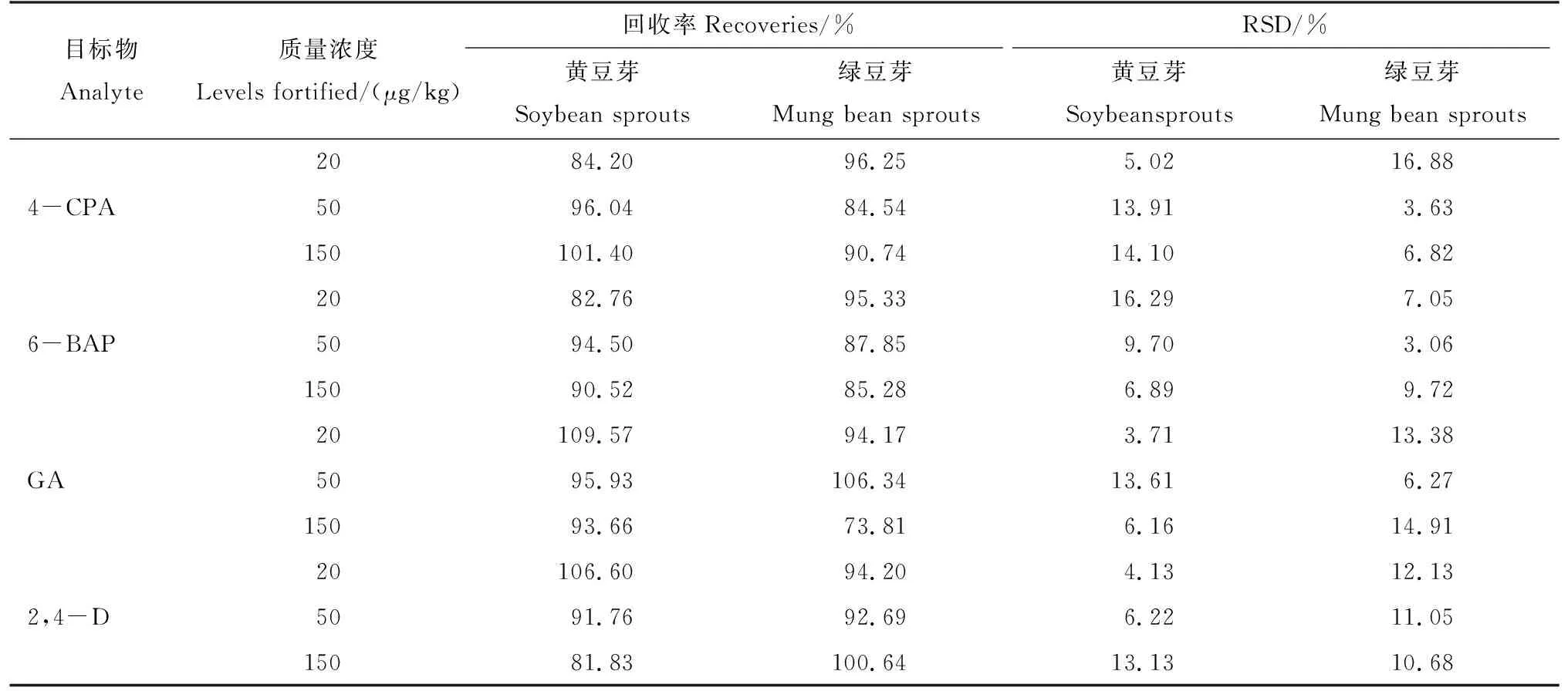
表2 加標(biāo)回收率與精密度(n = 6)Tab.2 Recoveries and precisions(n = 6)
2.5 實(shí)際樣品的檢測(cè)
采集超市和農(nóng)貿(mào)市場(chǎng)上豆芽產(chǎn)品50個(gè),其中黃豆芽28個(gè),綠豆芽22個(gè),應(yīng)用本試驗(yàn)建立的檢測(cè)方法進(jìn)行檢測(cè)。4-CPA,GA,2,4-D均未檢出,6-BAP有28個(gè)黃豆芽檢出,16個(gè)綠豆芽檢出,總檢出率為88%,但含量較低,均在LOD水平。說明6-BAP在豆芽生產(chǎn)中在普遍使用,也有可能是來源于環(huán)境污染,結(jié)果還尚有待進(jìn)一步確認(rèn)。
3 結(jié) 論
本研究建立了豆芽中4種典型植物生長(zhǎng)調(diào)節(jié)劑的液相色譜-串聯(lián)質(zhì)譜檢測(cè)方法,該方法具有樣品前處理簡(jiǎn)單,定量準(zhǔn)確,有足夠低的檢出限,能夠滿足監(jiān)測(cè)豆芽中關(guān)注度較高、豆芽中經(jīng)常被發(fā)現(xiàn)的4種植物生長(zhǎng)調(diào)節(jié)劑的需要。
猜你喜歡
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2020年12期)2021-01-18 06:57:46
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2020年12期)2021-01-18 06:57:46
兒童故事畫報(bào)(2019年5期)2019-05-26 14:26:14
海峽科技與產(chǎn)業(yè)(2016年3期)2016-05-17 04:32:12
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長(zhǎng)指南(2015年7期)2015-08-11 15:03:12
