氣相色譜-串聯質譜法同時測定水產干制品中 9種揮發性N-亞硝胺
2022-05-17 12:32:10陳紅波羅振玲
食品安全導刊 2022年10期
陳紅波,羅振玲,陸 正
(臺州市食品檢驗檢測中心,浙江臺州 318000)
N-亞硝胺是一類公認的致癌物質,廣泛存在于食品原料、飲用水、肉制品、水產制品中,結構通式為R2(R1)N-N=O,分為揮發性和非揮發性兩類,大多數揮發性N-亞硝胺是強致癌物質。含蛋白質的食品在加工過程中分解的胺類物質可與硝酸鹽/亞硝酸鹽生成N-亞硝胺,目前研究較多的有N-二甲基亞硝胺(N-dimethylnitrosamine,NDMA)、N-二乙基亞硝胺(N-diethylnitrosamine NDEA)、N-亞硝基甲 乙 胺(N-nitrosomethylethylamine,NMEA)、N-二丙基亞硝胺(N-dipropyl Nitrosamine,NDPA)、N-二丁基亞硝胺(N-dibutyl nitrosamine,NDBA)、N-亞硝基哌啶(N-nitroso Piperidine,NPIP)、N-亞硝基吡咯烷(N-nitrosopyrrolidine,NPYR)、N-亞硝基嗎啉(N-nitrosomorphine,NMOR)和N-亞硝基二苯胺(N-nitroso Diphenylamine,NDPhA)9種揮發性亞硝胺[1]。
水產干制品基質復雜,監測其中的N-亞硝胺含量,提取凈化方法是關鍵。現已報道的樣品前處理方法有水蒸氣蒸餾法[2]、液液萃取法[3]、固相萃取法[4]、QuEChERS[5]等方法。《食品安全國家標準 食品中N-亞硝胺類化合物的測定》(GB 5009.26—2016)采用的提取凈化方法為水蒸氣蒸餾、液液萃取和旋轉蒸發,提取方式煩瑣,過程復雜,耗時長,且蒸餾濃縮時揮發性目標物易損失。因此,優化或開發新的N-亞硝胺前處理方法是目前研究的熱點。N-亞硝胺的檢測方法有紫外和可見分光光度法、薄層色譜法、氣相色譜法、氣相色譜-熱能分析儀法、氣相色譜-質譜法、氣相色譜-質譜/質譜法和液相色譜-質譜/質譜法等。眾多方法中常用的是氣相色譜-質譜聯用法,但該方法在檢測食品中N-亞硝胺等小分子化合物時易受基質影響,產生較強的基質效應、本底信號高或假陰性等問題。而串聯質譜法可有效減少樣品基質的干擾,提高檢測結果的準確性和可靠性。
1 材料與方法
1.1 儀器與試劑
1.1.1 儀器
串聯三重四極桿氣相質譜聯用儀,Agilent 7890B+7000D型,美國安捷倫科技公司;渦旋震蕩儀,德國heidolph公司;離心機,ThermoFisher SCIENTIFIC。
1.1.2 試劑與耗材
乙腈,TEDIA,色譜純;二氯甲烷,TEDIA,色譜純;氯化鈉,天津市鼎盛鑫化工有限公司,分析純;硫酸,浙江漢諾化工科技有限公司,分析純;無水硫酸鎂,阿拉丁,分析純;PRiME HLB固相萃取小柱,Waters Oasis;EMR凈化管,美國安捷倫科技公司。
1.1.3 標準物質
9種亞硝胺混 標(NDMA(CAS 62-75-9)、NDEA(CAS 55-18-5)、NDPA(CAS 621-64-7)、NPYR(CAS 930-55-2)、NPIP(CAS 100-75-4)、NDBA(CAS 924-16-3)、NMEA(CAS 10595-95-6)、NDPhA(CAS 86-30-6)、NMOR(59-89-2))濃度分別為2 000 mg/L,1 mL,上海安譜實驗科技股份有限公司。
1.2 試驗樣品
魷魚絲、章魚干、墨魚干、水潺干、大黃魚鲞和蝦干。
1.3 試驗方法
1.3.1 水產干制品樣品前處理(1)水蒸氣蒸餾法。準確稱取20 g(精確至 0.01 g)試樣,加入20 mL水和10 g氯化鈉于蒸餾管中,充分混勻,檢查氣密性。在250 mL蒸餾燒瓶中加入 20 mL二氯甲烷及少量冰塊用以接收冷凝液,冷凝管出口伸入二氯甲烷液面下,并將平底燒瓶置于冰浴中,開啟蒸餾裝置加熱蒸餾,收集50 mL冷凝液后關閉加熱裝置,停止蒸餾。在盛有蒸餾液的平底燒瓶中加入5 g氯化鈉和3 mL的硫酸(1+3),攪拌使氯化鈉完全溶解。然后將溶液轉移至250 mL分液漏斗中,振蕩5 min,必要時放氣,靜置分層后,將二氯甲烷層轉移至另一平底燒瓶中,再用60 mL二氯甲烷分3次提取水層,合并4次二氯甲烷萃取液。將二氯甲烷提取液用適量無水硫酸鈉脫水后,進行旋轉蒸發,于40 ℃水浴上濃縮至5~10 mL改氮吹,并準確定容至1.0 mL,搖勻后測定。
(2)QuEChERS-EMR法。準確稱取10 g(精確至0.01 g)試樣,加入10 mL水浸泡30 min后加入 10 mL乙腈渦旋1 min,加入4 g無水硫酸鎂和1 g氯化鈉超聲15 min后渦旋2 min,離心5 min后取出上清液加入到EMR凈化管中,EMR凈化管事先已用5 mL水活化。凈化管渦旋10 min后離心5 min,取出上清液加入0.4 g氯化鈉和1.6 g無水硫酸鎂渦旋2 min后離心。取上清液4 mL氮吹至近干,加入 0.4 mL乙腈,經濾膜過濾至有內插管的進樣小瓶后測定。
(3)固相萃取法。準確稱取2 g(精確至0.01 g)樣品,加入10 mL乙腈后渦旋振蕩1 min,超聲 15 min,離心5 min。取5.0 mL上清液過PRIME HLB固相萃取小柱,凈化液氮吹至近干,加入 0.5 mL乙腈,經濾膜過濾后測定。
1.3.2 標準溶液配制
①標準儲備液A。取9種亞硝胺混標1 mL用乙腈稀釋至20.00 mL濃度為100 μg/mL,取0.1 mL該溶液用乙腈稀釋至10 mL,濃度為1 000 ng/mL。②標準儲備液B。取標準儲備液A 0.1 mL用乙腈稀釋至10 mL,濃度為10 ng/mL。③標準曲線溶液。分別準確移取標準儲備液A 1 μL、2 μL、5 μL、10 μL、 20 μL、50 μL、100 μL、200 μL和500 μL用乙腈稀釋至1.0 mL,濃度分別為1 ng/mL、2 ng/mL、5 ng/mL、10 ng/mL、20 ng/mL、50 ng/mL、100 ng/mL、200 ng/mL和500 ng/mL。
1.3.3 色譜和質譜條件
(1)色譜條 件。VF-WAXms柱(30 m×250 μm× 0.25 μm);高純氦氣;進樣量1 μL;脈沖不分流進樣模式;進樣口250 ℃;柱流速1.0 mL/min恒流;柱溫:50 ℃保持1 min,然后以15 ℃/min升溫至100 ℃保持1.5 min,再以5 ℃/min升溫至115 ℃保持1 min,再以40 ℃/min升溫至220 ℃保持8 min。
(2)質譜條件。掃描方式MRM模式;EI源電離,電壓70 eV;離子源溫度250 ℃;四極桿溫度(Q1和Q2)150 ℃;傳輸線溫度280 ℃。9種揮發性N-亞硝胺的MRM條件如表1所示。
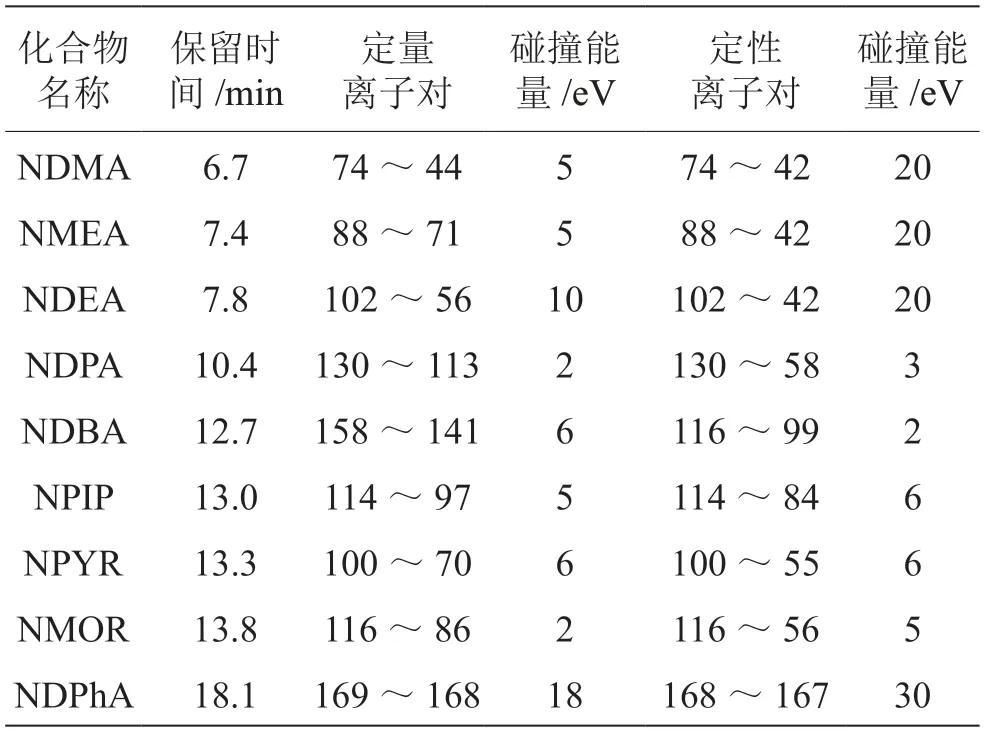
表1 9種揮發性N-亞硝胺的MRM條件
2 結果與分析
2.1 前處理方法選擇
水蒸氣蒸餾法是將《食品安全國家標準 食品中N-亞硝胺類化合物的測定》(GB 5009.26—2016)方法中的樣品稱樣量由200 g縮減至20 g,雖然樣品量減少能避免蒸餾不充分的缺點還能避免干擾和系統污染問題,但操作仍然復雜、步驟多、耗時長,有機溶劑消耗量大,N-亞硝胺在加熱濃縮過程中極易丟失,回收率不穩定。QuEChERS-EMR法樣品稱樣量小能避免干擾和系統污染問題,前處理過程與水蒸氣法相比比較簡便,快速有效,實現了N-亞硝胺在水產干制品樣品中的高回收率和穩定性,凈化效果較好。固相萃取法樣品稱樣量小能避免干擾和系統污染問題,所用的PRiME HLB固相萃取小柱無需活化可直接上樣,樣品溶液通過后,無需對小柱進行淋洗,操作簡單,快速有效。
選取魷魚絲試樣進行加標試驗(加標濃度 80 μg/kg),比較3種前處理方法的回收率,3種前處理方法均能實現對干制水產品中9種揮發性N-亞硝胺類化合物的檢測。HLB固相萃取法操作最簡單,快速有效,凈化效果較好,詳見圖1。
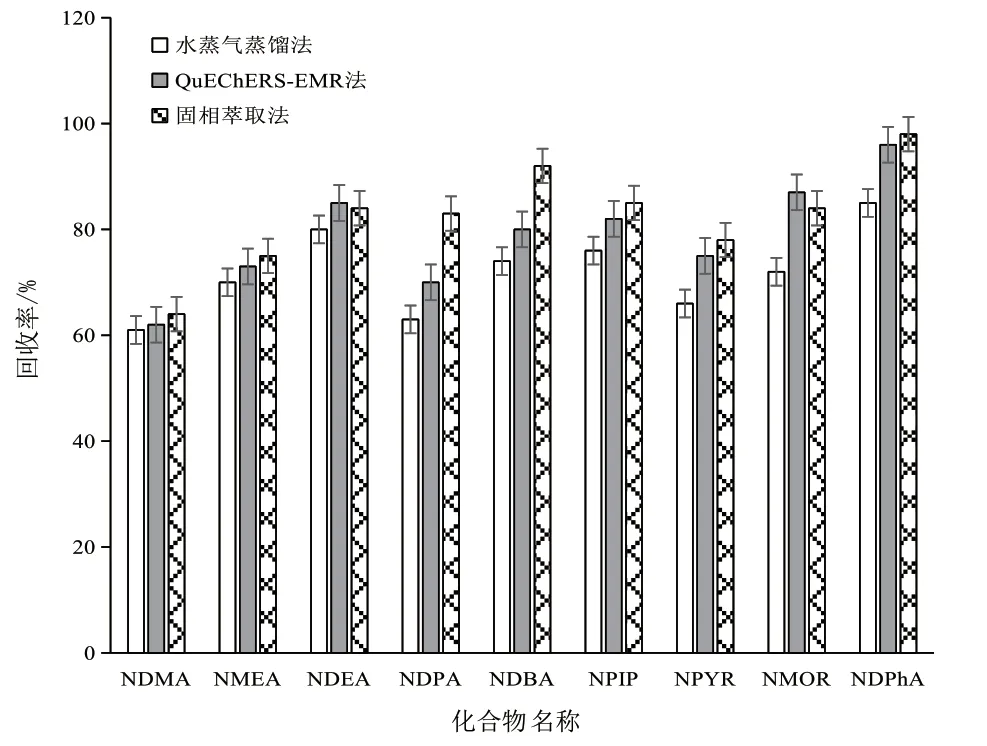
圖1 3種前處理方法的回收率圖
2.2 試驗條件選擇
揮發性N-亞硝胺類物質多用氣相色譜質譜法檢測,國家標準《食品安全國家標準 食品中N-亞硝胺類化合物的測定》(GB 5009.26—2016)中第一法也是采用氣相色譜質譜法,但化合物只經過一次篩選,本底信號高,基質影響較明顯,容易產生假陰性或假陽性結果。因此選用三重四極桿氣質聯用儀的MRM多反應離子監測模式。MRM多反應離子監測即母離子被選定,碰撞后,從形成的子離子中再次選定進行監測。經過2次篩選,MRM模式比SIM模式具有更強的降噪性能,因此采用MRM多反應離子監測模式可以極大地去除基質帶來的干擾。由圖2可知,樣品的總運行時間25 min,實現了一次進樣同時分析9種化合物,大大提高了分析效率。在MRM多反應離子監測模式下,各化合物經過定量離子對與定性離子對的提取,均實現了完美分離,且各化合物均無基質干擾,響應良好,能夠很好地實現水產干制品中9種N-亞硝胺類化合物的定性與定量分析。

圖2 9種揮發性N-亞硝胺的總離子流圖
2.3 方法線性及檢出限
取標準儲備溶液B 60 μL加入到2 g樣品中,按照固相萃取法進行樣品前處理,當各化合物的信噪比≥3時,該濃度即可確定為該化合物的檢出限濃度。由表2知,各化合物在1~500 μg/L(0.5~ 250.0 μg/kg)的濃度下,相關系數為0.999 6~0.999 9, 線性關系良好,檢出限為0.3 μg/kg,滿足檢測 需求。
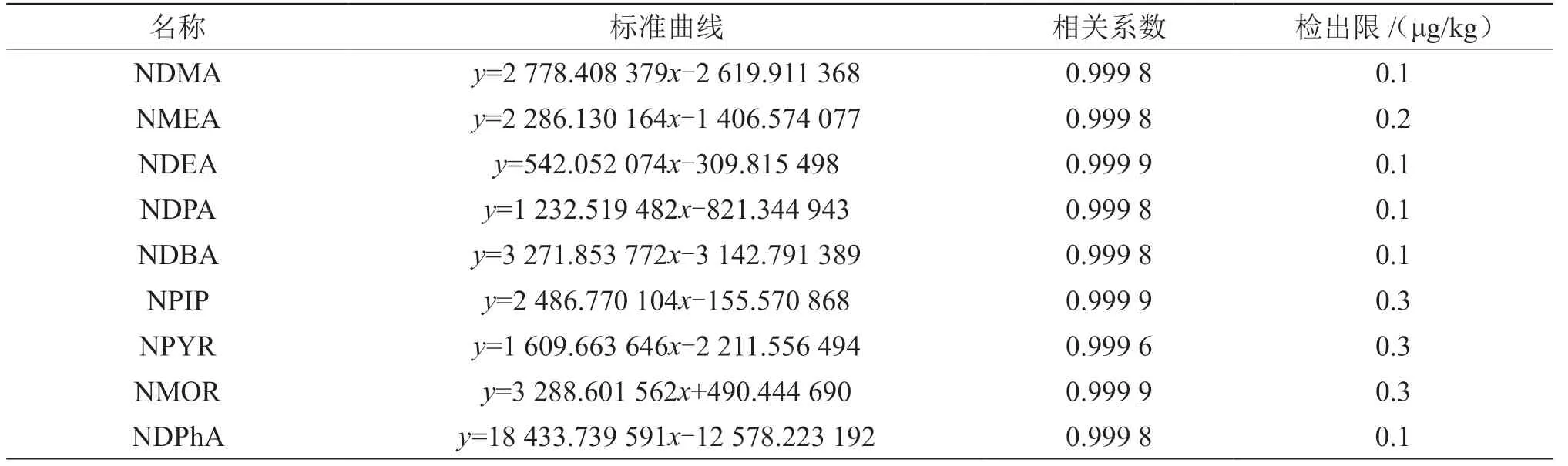
表2 9種N-亞硝胺類化合物的標準曲線及線性范圍
2.4 精密度與回收率
稱取2 g魷魚絲樣品18份,6份分別準確加入標準儲備液A 50 μL(對應加標濃度25 μg/kg),6份分別準確加入標準儲備液A 100 μL(對應加標濃度 50 μg/kg),6份分別準確加入標準儲備液A 200 μL(對應加標濃度100 μg/kg),按照固相萃取法進行樣品前處理,分別計算6份樣品的精密度和回收率。由表3可知,9種N-亞硝胺類化合物的加標回收率為70.1%~87.7%,精密度為1.6%~8.1%,回收率與精密度均滿足國家標準《實驗室質量控制規范 食品理化檢測》(GB/T 27404—2008)中相關規定,滿足檢測需求。
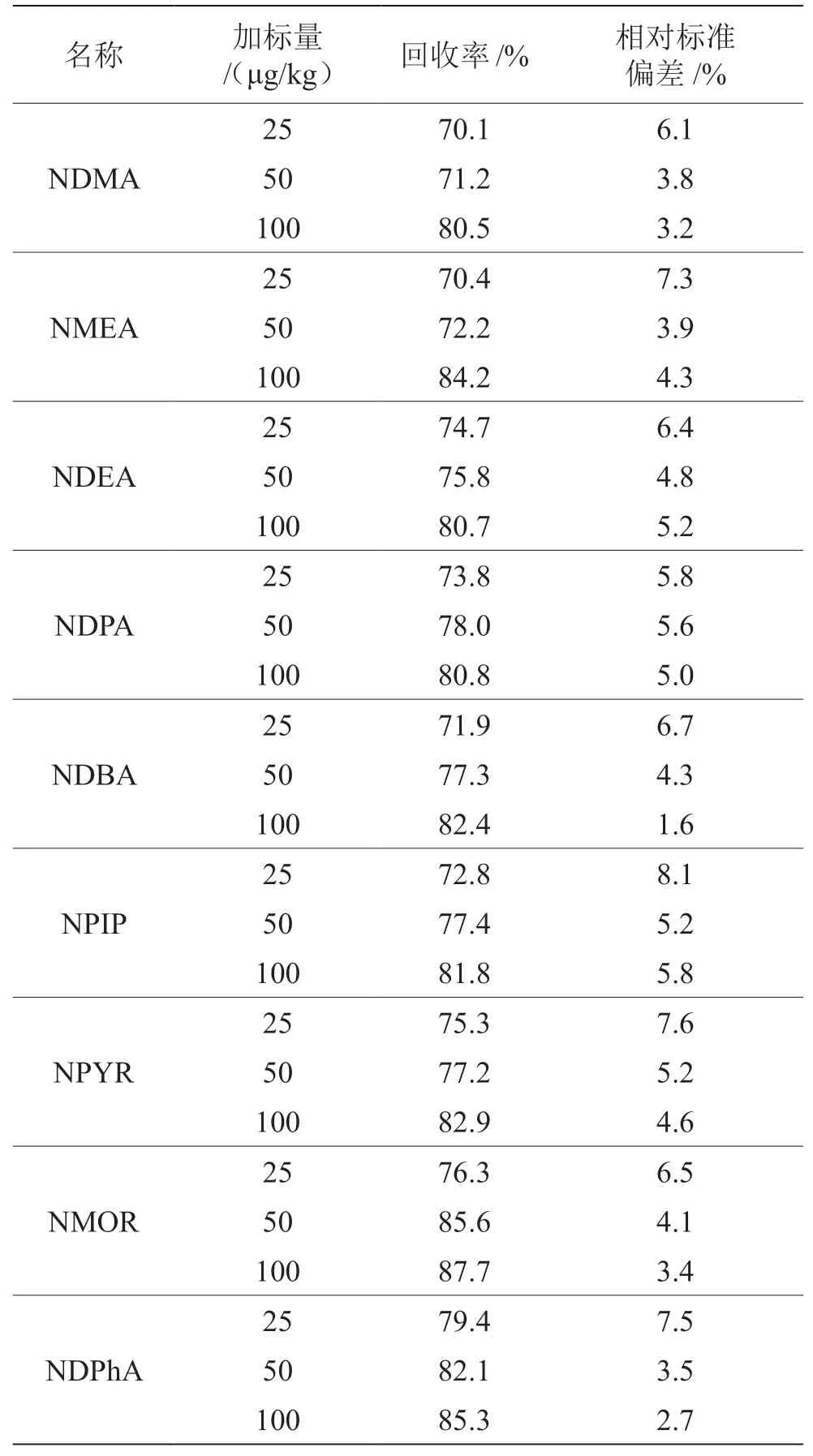
表3 回收率及精密度
2.5 樣品檢測
稱取粉碎混勻后的各試樣,選用固相萃取法進行樣品前處理,魷魚絲、章魚干、墨魚干、水潺干、大黃魚鲞和蝦干樣品中均有NDMA檢出,除NDMA外其他8種N-亞硝胺類化合物均未檢出。
3 結論
建立了水產干制品中9種揮發性N-亞硝胺的三重四極桿氣質聯用儀檢測方法,選用3種試樣前處理技術進行比較,3種前處理方法均能實現水產干制品中9種揮發性N-亞硝胺的檢測,凈化效果較好,回收率與穩定性均較高,都能滿足檢測需求,但HLB固相萃取法更簡便快速有效。選用固相萃取法進行實驗,在0.5~250 μg/kg質量濃度下,各化合物的響應值與濃度呈良好的線性關系(r>0.999 6),方法檢出限為0.3 μg/kg。取魷魚絲樣品分別進行3個不同濃度水平的加標實驗,回收率為70.1%~87.7%,精密度為1.6%~8.1%。取本地不同品種水產干制品進行檢測,各樣品中均檢出NDMA。方法前處理簡單,靈敏度高,適用于水產干制品中9種揮發性N-亞硝胺的同時檢測。
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
兒童故事畫報(2019年5期)2019-05-26 14:26:14
意林原創版(2016年10期)2016-11-25 10:28:30
海峽科技與產業(2016年3期)2016-05-17 04:32:12
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
