氮摻雜石墨烯原位生長碳納米管復合過渡金屬催化劑的制備及電催化性能
2022-05-23 04:54:10李生娟于沺沺李田成薛裕華
材料工程 2022年4期
關鍵詞:催化劑
張 婷,李生娟,吉 瑩,于沺沺,李田成,薛裕華
(上海理工大學 材料科學與工程學院,上海 200093)
近年來,可充電鋅空氣電池因安全性高、能量密度大、資源豐富、環境友好而被認為是一種很有前途的儲能裝置[1]。然而,在反應過程中,動力學緩慢、過電位大等缺點降低了鋅空氣電池空氣陰極上氧還原/氧析出(ORR/OER)的反應效率,從而限制其規模化應用。目前主要應用于ORR的貴金屬和OER的貴金屬氧化物催化劑分別有Pt,Pd和RuO2,IrO2等,但這些催化劑穩定性差、成本高、來源稀少、催化活性單一。因此,目前科研人員致力于開發高效、低成本、高性能的雙功能催化劑。一維碳納米管(CNTs)具有比表面積大、導電性好等優點,二維石墨烯(GO)具有大的比表面積、優異的導電性和良好的電化學穩定性等[2-3],將兩者結合可以得到具有優異導電性及電化學穩定性的三維復合結構。然而純碳材料催化活性較低,在進行催化反應時不可避免發生碳腐蝕[4],從而加劇催化劑的聚集。化學摻雜雜原子(如氮、磷、硼、硫等)是一種從本質上改變基體材料性質的有效方法,可以進一步提高其催化活性及穩定性。在元素周期表中,氮與碳相鄰,具有類似的原子半徑和電負性,因此,氮更容易融入碳晶格中,產生額外的孤對電子,加速電子的轉移,改善碳載體的電子性質,從而提高ORR活性。Liu等[5]報道了N摻雜碳納米管(NCNTs)中氮原子的摻雜可以有效地誘導碳材料中原子電荷密度和自旋密度的重新分布,并調節碳原子的O2吸附/解吸能,從而提高ORR活性。實驗和理論模擬研究表明,具有過渡金屬無機納米粒子的NCNTs顯示出比純CNTs更好的電化學性能[6-7]。盡管如此,這些材料的性能與商用Pt/C材料的性能還存在差距,限制了其在高能量密度器件中的應用。金屬有機骨架(MOFs)被認為是制備不同多孔結構電催化劑最有吸引力的前驅體或模板之一[8-10]。例如,利用MOFs材料制備摻雜雜原子的納米多孔碳[11]、碳包覆的金屬[12]以及它們的復合納米結構[13]。其中,鋅基沸石咪唑鹽骨架(ZIF8)是由2-甲基咪唑為有機配體,硝酸鋅提供金屬離子組成的有機框架結構,由于其富含C和N元素,所以是制備高性能氮摻雜碳材料的典型犧牲模板。然而MOFs材料本身導電性較差、高溫熱解后結構易坍塌且缺乏互連、石墨化程度低[4,13]等,使其在ORR反應中不能滿足電子快速轉移的要求,因此需要引入二次碳源(如氧化石墨烯,碳納米管等)或過渡金屬源調整其形貌來提高ORR活性。在氮摻雜碳過渡金屬復合材料中,溫度過高容易引起金屬的聚集,不利于活性位點的形成。在ZIF8中,二甲基咪唑配體中有明確的錨定位點,它可以作為多位點錨定來分散金屬,以抑制金屬納米粒子在熱解過程中的聚集[14]。此外,鋅的沸點為907 ℃[15],高溫下由于鋅的揮發,樣品會形成豐富的納米孔結構,有利于電子的傳輸。
本工作采用改良Hummers制備氧化石墨烯,以氧化石墨烯為載體,ZIF8為模板,尿素提供碳和氮源,無水FeCl3為金屬源,通過高溫熱解,ZIF8分解,在氮摻雜石墨烯片上原位生長碳納米管多孔復合結構,這種三維多孔結構有利于抑制金屬的聚集,暴露更多的活性位點,促進電子的快速傳輸,有助于電催化性能的提高。
1 實驗
1.1 催化劑的制備
本工作采用改良Hummers法進行催化劑的制備[16]。將5 g膨脹石墨粉和2.5 g NaNO3加入115 mL濃H2SO4中,在低于10 ℃的冰水浴中攪拌25 min,接著緩慢加入15 g KMnO4。25 min后升溫至35 ℃,攪拌45 min后加入230 mL去離子水,升溫至98 ℃,繼續攪拌45 min。然后加入350 mL去離子水,待冷卻至室溫后加入30 mL H2O2,攪拌30 min,接著加入30 mL HCl,攪拌30 min,得到混合溶液。將混合溶液靜置冷卻后用去離子水離心洗滌pH值接近6,最后超聲2 h,得到氧化石墨烯懸浮液(GO)。
將3 g尿素加入50 mL(1 mg/mL)氧化石墨烯懸浮液中,超聲2 h,得到均勻的棕色溶液。-80 ℃中冷凍12 h得到棕色固體,再置于冷凍干燥機中干燥48 h,得到尿素與氧化石墨烯復合的棕色粉末(N-GO)。
將4 g二甲基咪唑溶于25 mL甲醇中,攪拌10 min,得到二甲基咪唑溶液。取1 g Zn(NO3)·6H2O溶于10 mL甲醇中,攪拌10 min,得到Zn(NO3)·6H2O溶液。將二甲基咪唑溶液緩慢滴加到Zn(NO3)·6H2O溶液中,得到均勻的乳白色溶液,室溫下攪拌12 h,在8000 r/min轉速下離心20 min后,用甲醇洗滌3次,并在60 ℃下干燥5 h,得到ZIF8樣品。然后,取120 mg ZIF8,加入5 mL甲醇中,攪拌15 min,得到溶液A。60 mg的無水FeCl3加入5 mL甲醇,攪拌15 min,得到溶液B。將溶液A與溶液B混合攪拌12 h,在8000 r/min轉速下離心20 min后,用甲醇洗滌3次,并在60 ℃下干燥5 h,得到Fe/ZIF8樣品。
取1.25 g N-GO粉末與40 mg Fe/ZIF8,在研磨器中研磨混合30 min,在Ar氣氛中以3 ℃/min升溫到550 ℃,保溫2 h,接著升溫至900 ℃,保溫2 h,得到N/GO和Fe-ZIF8的復合材料,命名為N-GO@Fe/ZIF8-900。此外,為了探究N-GO@Fe/ZIF8-900的結構組成,分別將40 mg的Fe和ZIF8替換成等量的無水FeCl3和ZIF8,相同條件下,在900 ℃煅燒得到N-GO@Fe-900,N-GO@ZIF8-900作為對照樣品。
1.2 測試與表征
工作電極的制備:將5 mg催化劑溶于500 μL酒精中, 30 min超聲后加入470 μL去離子水,超聲30 min后加入30 μL Nafion溶液,最后超聲30 min形成均勻的油墨狀溶液。分別兩次取10 μL油墨狀溶液涂于玻碳電極上,形成直徑5 mm的催化劑膜涂層,催化劑負載量為0.404 mg/cm2。
采用X射線粉末衍射儀(D8 ADVANCE)對樣品進行物相分析,衍射源為CuKα(λ=0.15406 nm,掃描速率為3 (°)/min;拉曼光譜分析采用激光拉曼光譜儀(Horiba Lab RAM HR Evolution);采用掃描電子顯微鏡(FEI Quanta 450),透射電子顯微鏡(Tecnai G2 F30)觀察分析樣品的微觀形貌;使用比表面積分析儀(Micromeritics ASAP2020)進行比表面積和孔結構分析;電化學性能測量在電化學工作站(PINE AF)上進行;電池性能在電池測試系統(CT2001A)和電化學工作站(CHI760E)上進行。利用三電極體系,飽和甘汞電極作為參比電極(換算為可逆氫電極(RHE)),鉑片作為對電極,涂覆催化劑的旋轉環盤電極作為工作電極,在分別通入飽和O2和N2的0.1 mol的氫氧化鉀溶液中進行ORR和OER測試。ORR測試在0.2~1.2 V的電勢窗口內進行,掃描速率為10 mV/s,電極旋轉速率為400~2025 r/min。OER測試在1.2~2.0 V的電勢窗口內進行,掃描速率為10 mV/s,電極旋轉速率為1600 r/min。
鋅空氣電池的組裝:將15 mg的CNTs和5 mg催化劑置于50 mL的乙醇中超聲分散均勻,然后真空抽濾,形成一張均勻的膜,再用切片機切成直徑為8 mm的圓片作為催化層。催化劑負載量為0.2 mg/cm2,陰極從里向外依次為聚四氟乙烯膜、催化層、離子交換膜和泡沫鎳,清潔的鋅板和6.0 mol/mL的KOH分別用作陽極和電解液,用相同的方法將5 mg的20%Pt/C(質量分數)催化劑與15 mg的CNTs組裝成鋅空氣電池。
2 結果與討論
2.1 催化劑的組成與結構
圖1為N-GO@Fe/ZIF8-900樣品的合成示意圖。以石墨烯為基底,ZIF8和尿素提供碳和氮源,無水FeCl3為金屬源,通過高溫熱解形成氮摻雜石墨烯片上原位生長碳納米管結構。以氮摻雜石墨烯為基底,一方面提高導電性,另一方面碳氮形成C-N活性中心,同時ZIF8也為過渡金屬Fe提供更多的錨定位點。在煅燒過程中,石墨烯片上的含氧基官能團與Fe/ZIF8中的金屬離子有靜電相互作用[17],將Fe/ZIF8錨定在氮摻雜氧化石墨烯片上,在高溫下由于Fe的催化[18],ZIF8和N/GO原位生長出碳納米管結構。最終碳納米管與二維的石墨烯片形成氮摻雜石墨烯原位生長碳納米管三維多孔結構。
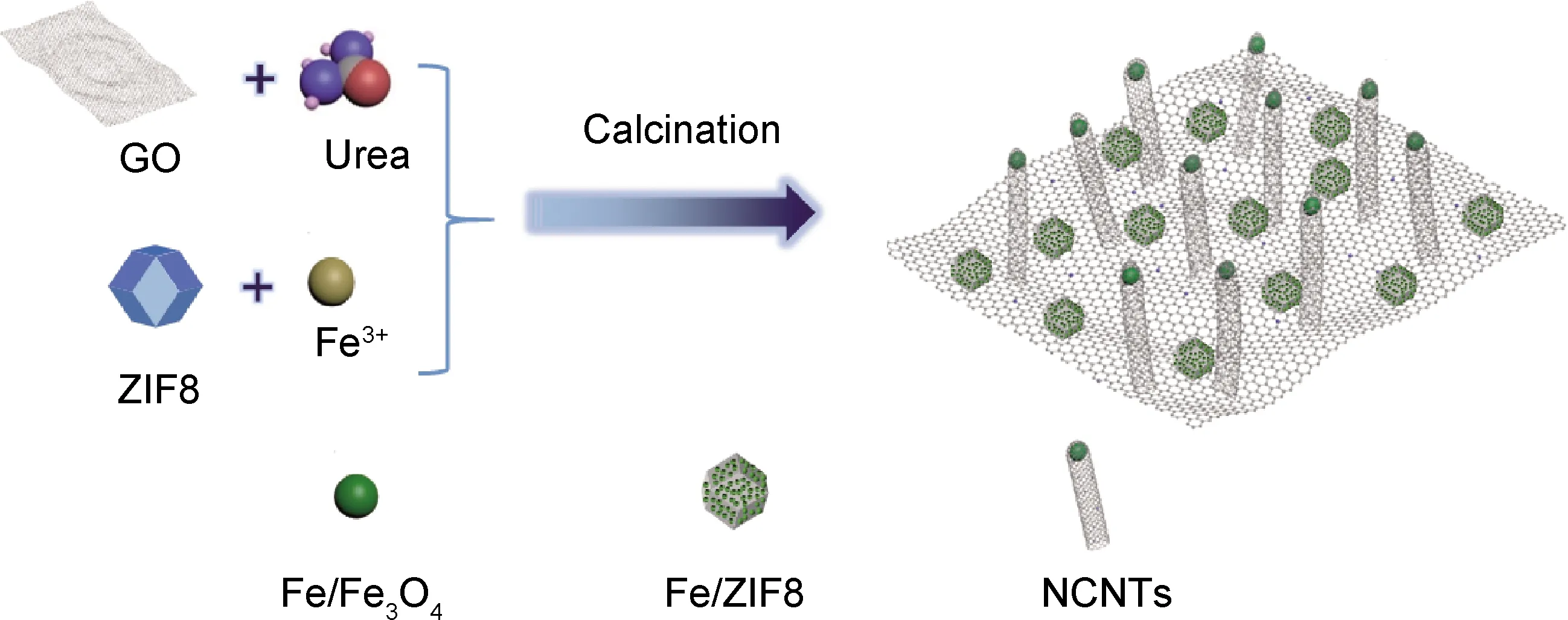
圖1 N-GO@Fe/ZIF8-900合成示意圖Fig.1 Schematic diagram of N-GO@Fe/ZIF8-900 synthesis
圖2為N-GO@ZIF8-900,N-GO@Fe-900,N-GO@Fe/ZIF8-900樣品的SEM圖。可知,煅燒后的ZIF8顆粒分布在氮摻雜石墨烯表面,未出現碳納米管(圖2(a));在石墨烯片上衍生出碳納米管結構,Fe/Fe3O4被包覆在金屬的頂端,直徑為(710±5) nm(圖2(b));在高溫煅燒過程中,金屬離子與氧化石墨烯片中的含氧官能團之間有著強相互作用,將Fe摻入ZIF8中形成Fe-ZIF8,并均勻錨定在GO納米片上(圖2(c))。以GO為基底,ZIF8為模板,尿素作為碳氮源,過渡金屬源作為催化劑,催化碳納米管生長[19-20],長成的碳管直徑為(70±5) nm,僅為N-GO@Fe-900碳管直徑的1/10左右。由于金屬催化碳納米管生長,碳納米管的直徑類似金屬顆粒的直徑。因此,通過將Fe-ZIF8與N/GO結合,衍生出直徑較小的金屬顆粒,有效抑制金屬的聚集。

圖2 N-GO@ZIF8-900(a),N-GO@Fe-900(b)和N-GO@Fe/ZIF8-900(c)的SEM圖Fig.2 SEM images of N-GO@ZIF8-900(a),N-GO@Fe-900(b) and N-GO@Fe/ZIF8-900(c)
圖3為N-GO@Fe/ZIF8-900樣品的TEM圖。可以看出,N/GO和Fe-ZIF8原位生長出碳納米管結構,碳納米管具有中空結構,含鐵納米顆粒(暗區)被包覆在碳納米管的頂端。由氮摻雜碳納米管(NCNTs)頂端的高分辨TEM放大圖可知,間距為0.21 nm的晶格條紋為立方鐵的(110)晶面,而間距為0.34 nm的晶格條紋為碳的(002)晶面,證實鐵納米粒子被限制在石墨碳納米管中。由N-GO@ Fe/ZIF8-900樣品中少量未分解完全的Fe/ZIF8透射電鏡圖可知,Fe/ZIF8主要以球形顆粒存在,含鐵納米顆粒顯示晶格條紋間距0.21 nm,為立方鐵(110)面,證明Fe成功摻入ZIF8框架中,形成氮摻雜石墨烯原位生長碳納米管的三維多孔結構。Fe與N的摻入可以形成很多Fe-Nx活性位點[21],這種三維多孔結構不僅有利于反應物在催化過程中的擴散,也有利于暴露更多的活性位點,從而促進電子的傳輸。

圖3 N-GO@Fe/ZIF8-900的TEM圖Fig.3 TEM images of N-GO@Fe/ZIF8-900
2.2 XRD,Raman和比表面積測試分析
圖4為N-GO@Fe-900,N-GO@Fe/ZIF8-900的XRD譜圖,Raman譜圖和比表面積測試圖。圖4(a)為N-GO@Fe-900,N-GO@Fe/ZIF8-900樣品的XRD譜圖,兩者的衍射峰與Fe(PDF No.87-0721),Fe3O4(PDF No.79-0418)和C(PDF No.75-1621)保持一致,表明金屬主要以Fe和Fe3O4的形式存在,碳主要以石墨碳的形式存在,說明氧化石墨烯在煅燒過程中被還原[22]。

圖4 N-GO@Fe-900,N-GO@Fe/ZIF8-900的XRD譜圖(a)和Raman光譜圖(b),N-GO@Fe/ZIF8-900的N2吸附/脫附等溫曲線(c)和孔徑分布曲線(d)Fig.4 XRD patterns(a) and Raman spectra(b) of N-GO@Fe-900 and N-GO@Fe/ZIF8-900,N2 adsorption/desorption isotherm(c) and pore size distribution curve(d) of N-GO@Fe/ZIF8-900
其中N-GO@Fe-900中Fe衍射峰的平均半高寬為0.427°,N-GO@ Fe/ZIF8-900中Fe衍射峰的平均半高寬為0.474°,峰寬較寬,說明晶粒尺寸較小,有利于活性位點的暴露[15,23],從而促進ORR/OER反應進程。圖4(b)為N-GO@Fe-900,N-GO@Fe/ZIF8-900的拉曼光譜圖,可以清楚地觀察到1340 cm-1(D峰)和1595 cm-1(G峰)處的兩個特征峰,其中D峰表示C原子晶格的缺陷,G峰表示C原子sp2雜化的面內伸縮振動。D峰和G峰的強度比為ID/IG,反映了碳材料的缺陷。相比于N-GO@Fe-900的ID/IG值(2.561),N-GO@Fe/ZIF8-900催化劑中ID/IG的值(2.837)較大,表明N-GO@Fe/ZIF8-900中存在更加豐富的缺陷位點,有利于反應過程中氧的吸附。通過N2吸附/脫附測試進一步表征900 ℃下N-GO@Fe/ZIF8樣品的比表面積和多孔結構,如圖4(c)所示,Ⅳ型等溫線線和磁滯(P/P0>0.45)處閉合,證實N-GO@Fe/ZIF8中存在大量的介孔結構,其中N-GO@Fe/ZIF8-900樣品的比表面積達到196.1 m2/g。圖4(d)的孔徑分布曲線顯示,N-GO@Fe/ZIF8-900樣品催化劑由介孔組成,孔徑為3.53 nm,孔容達到0.67 cm3/g,表明在900 ℃下尿素的分解(NH3CO(NH2)2=2NH3↑+HNCO)及鋅的揮發可以提供大的比表面積,有利于電子的傳輸。
2.3 XPS分析
采用X射線光電子譜(XPS)探究N-GO@Fe/ZIF8-900催化劑的成鍵和表面元素組成,如圖5所示。圖5(a)顯示N-GO@Fe/ZIF8-900催化劑中C,N,O,Fe和Zn的原子分數分別為85.82%,1.56%,6.62%,5.89%和0.11%,Zn的含量相當低,表明在煅燒過程中Zn基本揮發完全。圖5(b)為N-GO@Fe/ZIF8-900的C1s譜圖,其中285.3 eV的特征峰對應碳氮鍵[17,24-25],表明氮原子成功摻入碳骨架中。
圖5(c)顯示N-GO@Fe/ZIF8-900的N1s譜圖。N1s有四個特征峰,397.3 eV對應吡啶氮、398.2 eV對應吡咯氮、400.1 eV對應石墨氮、401.9 eV對應氧化氮,其中石墨氮和吡啶氮占總氮原子含量的百分比高達60.2%,如圖5(e)所示。氮摻雜到石墨烯結構中會導致電子分布不均,這些變化將促進氧的吸附和O—O鍵的解離,邊緣摻雜的石墨氮和吡啶氮降低了氧在相鄰碳原子上吸附的能壘,加速氧還原中電子的轉移,所以石墨氮和吡啶氮被認為是提高催化劑活性的主要基團[26]。圖5(d)為N-GO@Fe/ZIF8-900的Fe2p XPS譜圖,Fe2p有六個自旋軌道峰和一個衛星峰。Fe0對應718.9 eV和706.5 eV兩個峰,710.1 eV和723.3 eV的兩個峰被分配給Fe2+,Fe3+對應713 eV和732 eV的兩個峰,意味著Fe和Fe3O4存在,與XRD測試結果相符。圖5(e)中,N-GO@ Fe/ZIF8-900的Fe2p的Fe0∶Fe2+∶Fe3+的比例計算為10∶67∶23。豐富的Fe2+空位有利于形成Fe-Nx的活性中心[17],密度泛函理論計算證實Fe與N配位的相互結構有利于氧的吸附,Fe-Nx位點可以有效地裂解O—O鍵,在一定程度上提高ORR性能。此外,共存的Fe0,Fe2+和Fe3+氧化還原過程中電子的快速轉移明顯提高復合材料的導電性,供體-受體位點可以由鐵陽離子的價態交替提供,可逆地吸附/脫附氧提高ORR/OER活性。
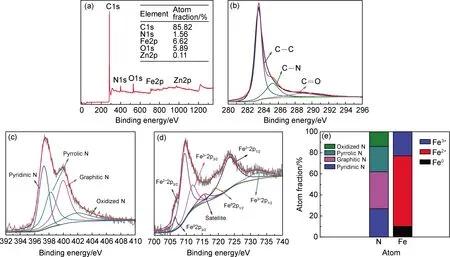
圖5 N-GO@Fe/ZIF8-900的XPS全譜圖(a),C1s(b),N1s(c),Fe2p譜圖(d)及不同類型N和Fe原子摻雜條形圖(e)Fig.5 XPS spectrum of N-GO@Fe/ZIF8-900(a),C1s spectra(b),N1s spectra(c),Fe2p spectra(d),and bar graph of different types of N and Fe atom doping(e)
2.4 ORR性能測試分析
圖6為不同催化劑的ORR性能測試圖。圖6(a)為不同催化劑樣品的LSV曲線,N-GO@Fe/ZIF8-900的半波電位達到0.885 V,優于N-GO@ZIF8-900(0.821 V),N-GO@Fe(0.849 V)和Pt/C(0.856 V),顯示出最優的氧還原性能。圖6(b)為N-GO@Fe/ZIF8-900在不同轉速下的LSV曲線,電流密度隨著轉速的增加而均勻增加。圖6(c)顯示N-GO@Fe/ZIF8-900催化劑樣品在不同電勢下利用K-L[15]方程計算獲得的氧還原反應過程中的電子轉移數n,當電勢在0.2~0.5 V時,催化劑N-GO@Fe/ZIF8-900的電子轉移數在3.67~3.98之間,表明在ORR過程中的電子轉移符合4e-轉移。圖6(d)為N-GO@Fe/ZIF8-900催化劑樣品在O2中穩定性測試曲線,可見持續運行16000 s后的電流密度仍保持在初始電流密度的87.6%左右,而Pt/C的電流密度保留率僅為70.6%,表明在堿性溶液中催化劑N-GO@Fe/ZIF8-900的穩定性優于商用Pt/C。
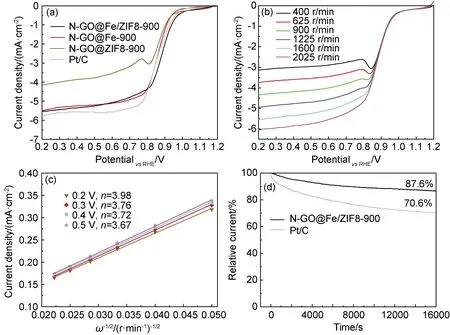
圖6 各催化劑在通入飽和O2的0.1 mol KOH溶液下的LSV曲線(轉速為1600 r/min,掃描速率為5 mV/s) (a),N-GO@Fe/ZIF8-900催化劑不同轉速下的LSV曲線(b),K-L曲線(c),I-T測試曲線(d)Fig.6 LSV curves of different catalysts in 0.1 mol KOH saturated with O2 (rotating speed:1600 r/min,scanning rate: 5 mV/s)(a),LSV curves(b),K-L curves(c),I-T test curves(d) of N-GO@Fe/ZIF8-900 catalyst under different speeds
2.5 OER性能測試分析
圖7為N-GO@Fe/ZIF8-900,Pt/C及IrO2的氧析出性能測試圖。圖7(a)的LSV曲線顯示,在10 mA/cm2的電流密度下N-GO@Fe/ZIF8-900樣品對應電位為1.811 V,優于貴金屬Pt/C (1.968 V),與IrO2(1.75 V)性能相當。圖7(b)為N-GO@Fe/ZIF8-900,Pt/C及IrO2的Tafel曲線,可以看出N-GO@Fe/ZIF8-900樣品的Tafel斜率為129.1 mV/dec,優于Pt/C(278.3 mV/dec),表明N-GO@Fe/ZIF8-900具有優異的雙功能催化活性。
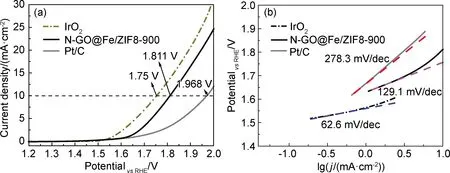
圖7 N-GO@Fe/ZIF8-900,Pt/C及IrO2樣品的LSV曲線(a)及Tafel曲線(b)Fig.7 LSV curves(a) and Tafel curves(b) of N-GO@Fe/ZIF8-900,Pt/C and IrO2 samples
2.6 電池性能測試分析
圖8為N-GO@Fe/ZIF8-900與Pt/C組裝成鋅空氣電池后的性能測試圖。圖8(a)為組裝成的鋅空氣電池在5 mA/m2條件下的循環穩定性測試對比圖。起始時N-GO@Fe/ZIF8-900樣品的電壓間隙(充電電壓-放電電壓)為0.949 V,而貴金屬Pt/C催化劑的電壓間隙為0.932 V。經過130 h的測試,兩者的電壓間隙分別為0.961,1.004 V,N-GO@Fe/ZIF8-900樣品的電壓間隙僅增加0.012 V,表明N-GO@Fe/ZIF8-900樣品具有更加優異的穩定性。圖8(b)為電流密度為10 mA/cm2時N-GO@Fe/ZIF8-900和Pt/C的比能量曲線。可以看出,N-GO@Fe/ZIF8-900的比能量達到886.2 mW·h·g-1,遠大于貴金屬催化劑Pt/C的比能量(791.04 mW·h·g-1)。圖8(c)為N-GO@Fe/ZIF8-900和Pt/C的極化曲線。N-GO@Fe/ZIF8-900的充放電電壓間隙小于Pt/C的電壓間隙,表明N-GO@Fe/ZIF8-900樣品組裝成鋅空氣電池時,具有優于Pt/C的充放電性能。圖8(d)為N-GO@Fe/ZIF8-900和Pt/C的功率密度曲線。N-GO@Fe/ZIF8-900的功率密度達到73.44 mW/cm2,優于貴金屬催化劑Pt/C的功率密度(57.12 mW/cm2),表明N-GO@ Fe/ZIF8-900樣品組裝的鋅空氣電池具有優異的電化學性能。

圖8 電流密度為5 mA/m2時組裝鋅空氣電池的循環穩定性測試曲線(a),電流密度為10 mA/cm2 時的比能量曲線(b),充放電極化曲線(c)及功率密度曲線(d)Fig.8 Cycle stability test curves of assembled zinc air battery at 5 mA/m2(a),specific energy curves at 10 mA/cm2(b),charge and discharge polarization curves(c) and power density curves(d)
3 結論
(1)通過直接熱解法構建一種氮摻雜石墨烯片上原位生長碳納米管多孔復合結構。N-GO@Fe/ZIF8-900催化劑與N-GO@Fe-900相比,有效抑制金屬的聚集。
(2)復合催化劑N-GO@Fe/ZIF8-900表現出良好的ORR/OER性能,在催化氧還原過程中,半波電位達到0.885 V,優于Pt/C(0.856 V)。在催化氧析出過程中,10 mA/cm2的電流密度下N-GO@Fe/ZIF8-900樣品對應電位為1.811 V,優于貴金屬Pt/C(1.968 V),與IrO2(1.75 V)性能相當,表明N-GO@Fe/ZIF8-900具有良好的雙功能催化活性。
(3)復合催化劑N-GO@Fe/ZIF8-900組裝成鋅空氣電池,具有優于Pt/C的循環穩定性,比能量和功率密度分別達到886.2 mW·h·g-1和73.44 mW/cm2,高于貴金屬Pt/C的比能量(791.04 mW·h·g-1)和功率密度(57.12 mW/cm2),有望成為在可充電鋅空氣電池中貴金屬催化劑的替代品。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
