褪黑素基于線粒體保護治療心肌梗死的研究進展
2022-05-31 04:24:34張子睿呂婷婷李尚孫嵐
江蘇大學學報(醫(yī)學版) 2022年3期
關鍵詞:融合
張子睿, 呂婷婷, 李尚, 孫嵐
(中國醫(yī)學科學院北京協(xié)和醫(yī)學院藥物研究所藥物篩選中心,北京 100050)
近年來,急性心肌梗死早期的再灌注治療顯著提高了患者的生存率,然而心肌梗死患者的預后仍不理想。當缺血區(qū)域再灌注時,氧氣的突然恢復會造成氧化應激,導致心肌代謝紊亂與結構受損[1],是心肌缺血再灌注損傷的主要機制。而在心肌梗死后期心臟修復的過程中,心室容易發(fā)生不良重構,部分心肌超負荷與功能心肌丟失,最終導致心肌梗死后的心力衰竭[2]。
心臟需要大量ATP以維持收縮功能和離子轉運,因此線粒體占心肌細胞體積高達30%,以持續(xù)且快速地供應能量[3]。經大量研究證實,線粒體在心肌缺血再灌注與心肌梗死后心室重構中都發(fā)揮著至關重要的作用,保護線粒體功能和維持線粒體穩(wěn)態(tài)能夠改善心肌代謝功能,減少心肌梗死面積并抑制心室不良重構[4]。
褪黑素(melatonin)是一種由哺乳動物松果體分泌的吲哚類激素,在線粒體中有著較高的濃度,其對線粒體的作用已在先前的研究中得到廣泛探索。褪黑素已被證明可以改善心肌梗死誘導的線粒體氧化應激、膜電位變化、線粒體動力學和線粒體自噬的紊亂[5],并作為一種有吸引力的心肌梗死治療策略引起了人們的關注。
1 褪黑素-線粒體軸降低心肌梗死損傷
研究表明,褪黑素能夠通過抑制心肌梗死后線粒體通透性轉換孔(mitochondrial permeability transition pore,mPTP)開放[6]與抑制線粒體活性氧生成[7]以降低線粒體氧化應激,維持線粒體膜電位穩(wěn)定性并提升ATP產量,抑制心肌梗死后的線粒體裂變升高并增強線粒體融合以維持線粒體動力學的動態(tài)平衡[8],從而抑制線粒體自噬,保護線粒體功能,降低心肌缺血再灌注損傷并抑制心肌梗死后不良重構(圖1)。
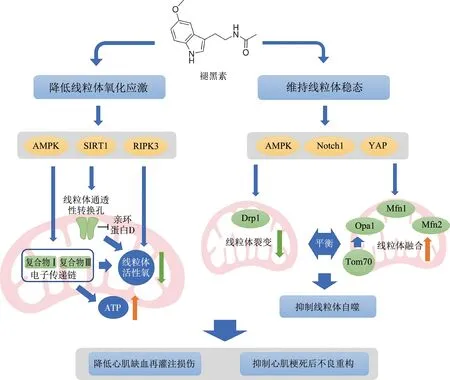
AMPK:腺苷酸活化蛋白激酶;SIRT1:沉默信息調節(jié)因子1;RIPK3:受體相互作用蛋白3;YAP:Yes相關蛋白;Drp1:線粒體動力相關蛋白1;Opa1:視神經萎縮蛋白1;Mfn1:線粒體融合蛋白1;Mfn2:線粒體融合蛋白2
1.1 褪黑素降低線粒體活性氧
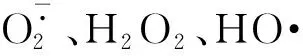
褪黑素作為一種天然的強效抗氧化劑,能夠提高缺血心肌線粒體中復合物Ⅰ和復合物Ⅲ的活性,加速電子通過ETC的速度并減少了電子的泄漏,抑制活性氧的產生,降低氧化應激產生的心肌損傷[10]。褪黑素清除活性氧并降低心肌缺血再灌注損傷的治療作用已得到多方驗證,其具體機制仍在探究中。褪黑素能夠通過上調SIRT3,減輕H2O2誘導的H9c2細胞死亡和細胞內活性氧聚集,并在小鼠缺血再灌注模型中以SIRT1依賴性激活SIRT3信號通路,降低線粒體中的酶促超氧化物清除劑SOD2的乙酰化,減少線粒體活性氧的產生[7]。腺苷酸活化蛋白激酶(adenosine 5′-monophosphate-activated protein kinase,AMPK)是一種重要的心臟能量傳感器,能夠在生理病理狀態(tài)下維持線粒體能量穩(wěn)態(tài)。褪黑素治療缺血再灌注心肌顯著增強了AMPK的磷酸化,降低線粒體的活性氧生成,增加了Nrf2的核轉位,維持氧化反應平衡[11],并通過激活AMPK/mTOR通路降低心肌缺血再灌注后的微血管損傷[12]。
線粒體解偶聯蛋白(uncoupling protein,UCP)的激活通常對線粒體功能產生有益作用,包括平衡膜電位、加速電子傳輸、減少活性氧形成和細胞的氧化損傷。Aslan等[20]在大鼠缺血再灌注模型中敲除UCP3顯著增加了活性氧的生成,梗死面積擴大2倍,褪黑素的使用有效提高了UCP3的表達量,并對缺血后損傷有著明顯的保護作用。
1.2 褪黑素抑制mPTP的開放
mPTP是位于線粒體內外膜之間的非選擇性通道,具有維持線粒體膜電位和細胞內外離子平衡的作用。當心肌缺血時mPTP關閉,而當再灌注發(fā)生時,氧氣供給突然恢復導致活性氧累積和Ca2+超載,促使mPTP開放。病理性的mPTP開放會導致累積的活性氧爆發(fā)性外流引起氧化應激、膜電位消散抑制ATP生成和凋亡因子大量外流細胞質引起細胞凋亡,是心肌缺血再灌注造成心肌損傷的重要原因[21]。研究表明[22],褪黑素治療心肌缺血再灌注時,心肌中mPTP的開放程度會明顯下降,說明褪黑素很有可能通過抑制mPTP的開放從而降低缺血再灌注帶來的損傷。mPTP的開放在結構上受親環(huán)蛋白D(cyclophilin D,CypD)的磷酸化調控,Zhou等[6]發(fā)現在心肌損傷12 h前腹腔給藥褪黑素(20 mg/kg)能夠抑制受體相互作用蛋白激酶3(ripk3)和絲氨酸蛋白磷酸酶5(PGAM5)的上調,抑制CypD磷酸化,從而使mPTP開放正常化。除此之外,線粒體電壓依賴陰離子通道將細胞質己糖激酶2與線粒體緊密結合,通過抑制Bax/Bad與線粒體的相互作用來防止 mPTP 開放,褪黑素通過激活AMPK促進己糖激酶2遷移到線粒體,使己糖激酶2與線粒體結合從而抑制mPTP的開放,并改善了膜電位[23]。心磷脂是mPTP的重要組成部分,心磷脂或線粒體連接蛋白 43(mitochondrial connexin 43,mtCx43)信號通路調節(jié)線粒體膜流動性,可在氧氣利用率降低的條件下維持心肌能量。Petrosillo等[24]使用過氧化叔丁醇(t-BuOOH)處理大鼠心臟中的線粒體使心磷脂過氧化,發(fā)現使用褪黑素可防止和逆轉過氧化的進行從而保護心磷脂,10 μmol/L褪黑素基本可以完全阻止Ca2+/t-BuOOH引起的膜電位降低,從而防止細胞色素c從膜上脫離和 mPTP的開放,同時褪黑素不影響線粒體對Ca2+的累積。mPTP開放是再灌注損傷的后期階段,在H9c2細胞上的研究表明,褪黑素后處理保護H9c2細胞免受缺血再灌注損傷的治療時間窗較大,再灌注開始后15~30 min內給藥都能有效降低mPTP的開放[25]。
1.3 褪黑素調節(jié)線粒體裂變與融合
線粒體是一種動態(tài)的細胞器,能夠通過線粒體裂變和融合不斷改變形態(tài),以此控制線粒體質量并調控線粒體自噬。研究發(fā)現線粒體裂變能將線粒體分裂成更小的形態(tài),產生適當大小的線粒體碎片以能夠被自噬體吞噬;反之線粒體融合能使兩個線粒體融合成一個更長的線粒體并抑制線粒體自噬。線粒體融合與裂變蛋白在心臟中廣泛表達,線粒體融合主要由外膜融合蛋白 Mfn1 和 Mfn2 以及內膜融合蛋白Opa1完成,線粒體裂變主要由Drp1 和 Fis1 蛋白完成[26]。
在正常生理過程中,線粒體融合與裂變處于動態(tài)平衡的狀態(tài)。而在心肌缺血、心肌細胞受損以及缺血再灌注時,由于線粒體裂變被過度激活,而線粒體融合能夠使破碎的線粒體重新融合,因此抑制線粒體裂變與促進線粒體融合是降低心肌梗死損傷的可能機制。褪黑素能夠調節(jié)線粒體裂變與融合以維持線粒體的穩(wěn)態(tài)[27]。心肌細胞的線粒體裂變主要由Drp1調控。在大多數情況下,褪黑素能夠降低Drp1的表達從而減少線粒體裂變,并降低Drp1的絲氨酸616磷酸化,以抑制線粒體Drp1移位[28],從而起到減少心肌細胞凋亡,改善線粒體功能和心臟功能[29],抑制血管鈣化的功效[30]。在心肌缺血再灌注時,心肌中Drp1被顯著激活,而再灌注時使用褪黑素治療也被證明能夠降低磷酸化Drp1的表達[8]。褪黑素抑制Drp1的具體機制仍在探究中,Chen等[30]在主動脈粥樣硬化的實驗中證明褪黑素對Drp1的抑制作用基于對AMPK的激活。但在心肌梗死治療中更加深入的機制研究仍然缺乏。
線粒體融合分為外膜融合與內膜融合,主要由內膜融合蛋白Opa1與外膜融合蛋白Mfn1和Mfn2介導。Opa1與Opa1相關的線粒體融合有助于維持心肌缺血再灌注應激條件下的線粒體穩(wěn)態(tài),褪黑素已被證明能夠通過調控并激活Opa1的表達以減輕心肌缺血再灌注損傷。Zhang等[31]使用心臟特異性敲除Opa1模型小鼠,證實褪黑素對心肌缺血再灌注時炎癥反應、鈣超載與氧化應激的抑制作用以及對線粒體融合的激活作用都需要Opa1介導,在H9c2缺氧復氧模型中也證明了Opa1在褪黑素治療過程中的必要性[32]。褪黑素介導Opa1激活的機制有多種說法,研究發(fā)現AMPK、ERK和Yes相關蛋白(Yes-associated protein,YAP)可以調節(jié)Opa1的表達以減少缺氧誘導的心肌細胞死亡[33],更加深入的機制研究也在逐步開展中。如Ma等[34]發(fā)現在小鼠再灌注損傷模型中,褪黑素能夠逆轉由于心肌損傷導致的YAP和Opa1下調,且使用YAP拮抗劑維替泊芬后抑制了褪黑素導致的Opa1上調;Zhang等[31]證明AMPK是Opa1的上游信號,褪黑素能夠促進AMPK的磷酸化,并通過AMPK-Opa1軸抑制細胞色素c的釋放,從而抑制線粒體的凋亡。
線粒體融合蛋白Mfn1和Mfn2位于線粒體外膜上,是外膜融合所必需的蛋白[26]。褪黑素能夠顯著升高心肌細胞中Mfn1和Mfn2的表達,逆轉缺氧導致的H9c2細胞中Mfn1和Mfn2的降低,增強了缺氧心肌的線粒體融合[34]。據報道Mfn2缺失會顯著惡化小鼠心肌缺血引起的心臟損傷,但褪黑素治療后能逆轉這一效果,減少了缺血后的心肌細胞凋亡與心肌纖維化,抑制了心肌梗死后心肌的不良重塑[2]。Notch1是褪黑素調控Mfn2的關鍵因子,能通過調控線粒體自噬關鍵通路Pink1/Parkin維持線粒體穩(wěn)態(tài)[27],并促進Mfn2和Opa1的表達,抑制過度的線粒體裂變[35]。干擾Notch1降低了褪黑素的治療效果,且使用褪黑素受體抑制劑luzindol處理后證明了Notch1上調Mfn2依賴于褪黑素受體。褪黑素處理增加了線粒體中Notch1、Notch1胞內結構域、Hes1、Bcl-2的表達和p-Akt/Akt比值,降低了凋亡因子Bax和Caspase-3的表達[36]。Notch1也被證明通過上調RBP-Jk激活Mfn1并抑制Drp1,從而抑制心肌缺血再灌注損傷,并提高線粒體膜電位恢復線粒體產能[37]。但有趣的是,有報道顯示Mfn1和Mfn2缺失反而在心肌細胞應對損傷時具有正面效果,Mfn2缺失的成人心肌細胞對Ca2+超載耐受能力更強,在再灌注損傷后表現出更好的恢復能力[38];而Mfn1敲除會使線粒體呈小球狀但不影響線粒體正常的呼吸功能,反而能夠提高線粒體對活性氧誘導的功能障礙的耐受性,對mPTP開放導致的線粒體功能障礙具有更大的抵抗力[39]。這與之前所述的研究結果相矛盾,有待進一步研究與討論。
1.4 褪黑素靶向線粒體轉運蛋白
線粒體轉運蛋白是大多數前體蛋白進入線粒體的入口,負責識別細胞質基質中的前體蛋白,包括Tom復合體、Tim復合體等。Tom70是Tom中的重要組成部分,缺血或缺氧損傷會降低心肌細胞中的Tom表達。心肌肥大是心肌梗死后不良重構中的重要標志之一,能夠進一步引發(fā)心力衰竭。研究發(fā)現在病理性心肌肥大中Tom70下調,且在體內外實驗中敲低Tom70均能夠誘導顯著的病理性心肌肥大。進一步探究證明了Tom70能夠通過調控Opa1促進線粒體融合,對維持線粒體形態(tài)至關重要,且用腺病毒介導的Tom70過表達完全消除了異丙腎上腺素誘導的心肌肥大[40]。Tom70在心肌缺血再灌注中的作用也被證實,Tom70在缺血再灌注30 min后表達顯著降低,Tom70的缺乏會降低線粒體鈣離子攝入蛋白1(MICU1)的定位并增加Ca2+含量與線粒體碎片化,額外補充Tom70能夠減少梗死面積,抑制心肌肌鈣蛋白T(cTnT)泄漏[41]。Pei 等[42]研究了 Tom70 在小鼠體內永久冠狀動脈結扎心肌梗死模型和培養(yǎng)的新生鼠心肌細胞缺血模型中的作用,結果表明褪黑素預處理 (10 mg/kg/d) 上調PGC-1α 表達并減輕心肌損傷,但在 Tom70缺乏的小鼠中褪黑素的作用被抑制了,且褪黑素對Tom70的調控為褪黑素受體依賴性。
1.5 褪黑素抑制心肌梗死后的心肌細胞焦亡
焦亡是近年來發(fā)現的一種細胞程序性死亡方式,主要由Gasdermin家族中的GSDMD和GSDME調控,其能夠誘導細胞凋亡向細胞焦亡轉換,并誘導炎癥的發(fā)生。GSDMD誘導的心肌細胞焦亡是心肌缺血再灌注損傷中的關鍵因素,缺血再灌注激活caspase-11、切割GSDMD為GSDMD-N并誘導凋亡,因此抑制GSDMD是降低心肌缺血再灌注損傷的有效手段[43]。研究發(fā)現,褪黑素能夠抑制GSDMD的表達,從而抑制脂多糖誘導的心肌細胞焦亡[44]。線粒體活性氧被證明能夠激活NLRP3炎癥小體的表達[45],而Wen等[46]在小鼠心肌梗死模型中發(fā)現,褪黑素通過TLR4/NF-κB通路抑制NLRP3炎癥小體誘導的心肌細胞焦亡。除此之外,GSDME誘導的焦亡與線粒體有著重要的關系,GSDME-N能夠透化線粒體膜,使其釋放細胞色素c并促進凋亡[47],且線粒體mPTP促使Apaf-1與caspase-4結合為新型蛋白復合物,并通過此復合物激活caspase-3-GSDME導致快速的細胞焦亡[48]。但目前尚缺乏褪黑素針對GSDME誘導的細胞焦亡相關研究,有待更深入的機制探索。
2 結論
褪黑素作為一種天然的強效線粒體保護劑,對心肌梗死的急性期治療和后期心臟重構的改善都有明顯的優(yōu)勢和潛力。除了本文關注的線粒體保護作用外,研究證明褪黑素還具有抑制凋亡和抗炎的作用[49],對調節(jié)血脂、血小板功能和微循環(huán)有著潛在的益處。褪黑素作為一種內源性分子,具有較高的生物安全性。但同時褪黑素具有強晝夜節(jié)律性,在體內具有完整的調節(jié)與代謝機制,外源性補充褪黑素難以達到穩(wěn)定的血藥濃度。臨床研究發(fā)現,與健康者相比,急性心肌梗死患者心臟中的褪黑素水平在白天沒有較大的差異,但在夜晚會顯著降低[50]。因此,外源補充褪黑素應該更多考慮晝夜節(jié)律的影響,其給藥方式、給藥時間與給藥劑量需要進一步的臨床探究。
猜你喜歡
中學生數理化·中考版(2022年8期)2022-06-14 06:55:24
數學年刊A輯(中文版)(2022年4期)2022-02-16 08:17:34
今日農業(yè)(2021年19期)2022-01-12 06:16:36
中老年保健(2021年11期)2021-08-22 03:15:44
無線電通信技術(2021年4期)2021-07-13 08:58:28
無線電通信技術(2021年3期)2021-06-08 03:33:48
中學生數理化(高中版.高考數學)(2021年1期)2021-03-19 08:28:38
無線電工程(2020年11期)2020-10-29 01:25:46
現代出版(2020年3期)2020-06-20 07:10:34
福利中國(2015年4期)2015-01-03 08:03:38
