嵌入型Ni@S1催化劑的制備及其CH4-CO2重整反應性能
2022-06-01 01:35:22蓋希坤朱繼成駱美宇馬清祥張良佺
潔凈煤技術 2022年5期
關鍵詞:催化劑
蓋希坤,楊 丹,朱繼成,駱美宇,馬清祥,邢 闖,呂 鵬,張良佺
(1.浙江科技學院 生物與化學工程學院 浙江省農產品化學與生物加工技術重點實驗室,浙江 杭州 310023;2.寧夏大學 省部共建煤炭高效利用與綠色化工國家重點實驗室,寧廈 銀川 750021)
0 引 言
隨著全球氣候變暖問題日益突出,CO2的減排、捕獲和轉化利用技術成為研究熱點[1-2]。CH4是天然氣的主要成分,具有清潔、高效、無污染的特性[3-4]。但同時也是溫室氣體。CH4-CO2重整(DRM)反應能夠將2種溫室氣體CH4和CO2轉化為合成氣,作為羰基合成和費托合成等反應的原料。1991年Aschcroft發表了關于CH4-CO2重整反應的研究引發了廣泛關注[5]。30多年來,國內外學者對該反應催化劑制備、反應機理、催化劑失活抑制等方面進行了大量研究[6-8]。低成本、高活性和高穩定性的催化劑依然是影響該反應實現工業化應用的主要因素。
DRM反應催化劑的活性組分為VIII族過渡金屬(除Os),主要包括兩大類:貴金屬(Ru、Rh、Ir、Pd、Pt)和非貴金屬(Ni、Co、Cu、Fe)。貴金屬雖然催化活性和抗積碳性能好,但資源有限、價格昂貴,應用有限。非貴金屬中Ni具有很強的催化活性,且價格低廉,工業應用前景巨大[9-10]。然而,Ni基催化劑存在高溫下易燒結和積碳導致失活的缺點,為解決此問題,研究者對Ni基催化劑進行了大量研究[11-14]。Ni顆粒大小對DRM反應的催化性能影響顯著,減小Ni顆粒能有效提高催化劑的抗積碳性能[15-16]。但在高溫條件下,小顆粒易遷移形成大顆粒,導致催化劑活性下降。研究者提出了多種避免Ni顆粒聚集的策略,如制備核殼催化劑和利用介孔硅材料的約束作用等。PENG等[17]合成了一種M-Ni@SiO2微孔催化劑,800 ℃反應100 h后,催化劑上未檢測到積碳,但Ni顆粒尺寸從4.3 nm增大到5.2 nm。LI等[18]報道了一種通過降解AlN得到的Ni@Al2O3/AlN催化劑,由于Al2O3覆蓋層的物理限制以及Ni和AlN的強相互作用,反應后Ni@Al2O3/AlN催化劑上未檢測到積碳。但由于Al2O3部分覆蓋了Ni活性位點,導致Ni@Al2O3/AlN的催化活性降低。近年來,金屬鑲嵌分子篩的催化劑在DRM中得到廣泛關注[19-22]。WANG等[19]采用簡單的一鍋水熱法制備了同時具有物理化學雙約束微孔封裝型Ni@S-2-E催化劑,Ni活性金屬成功嵌入分子篩S-2-E中,且NiNPs僅為2.6 nm左右,與負載型Ni/S-2催化劑相比,抗燒結性能和抗積碳性能均得到顯著提高。在分子篩的工業化合成中,水熱法應用最多,水熱過程會消耗大量溶劑而且產生大量廢水。2012年肖豐收課題組[23]提出了無溶劑硅鋁酸鹽合成法,通過簡單固相研磨將活性金屬嵌入分子篩,實現活性金屬的限域,該方法與傳統水熱法相比具有廢水排放少、簡單快捷和收率高等優點。
筆者采用研磨-晶化法在晶體成核過程中嵌入Ni顆粒,合成了Ni質量分數分別為5%、15%的原位微孔晶體的Ni@Silicalite-1(簡稱S1)催化劑,并應用于DRM反應,考察嵌入型結構對DRM反應催化性能的影響。
1 試 驗
1.1 催化劑制備
浸漬法制備負載型5Ni/Q10、15Ni/Q10催化劑:將一定量在烘箱中過夜干燥的Q10(孔徑10 nm的無定形SiO2)作為載體放入圓底燒瓶中。取適量硝鎳溶于去離子水,再將其加入圓底燒瓶進行旋蒸,保持溫度在70 ℃并不斷攪拌,使硝酸鎳均勻負載在載體上,得到Ni質量分數分別為5%、15%的Ni/Q10催化劑前驅體。抽真空3 h,100 ℃恒溫干燥12 h,550 ℃空氣氛圍中焙燒4 h,冷卻至室溫后造粒成0.850~0.425 mm(20~40目),儲藏備用。
研磨-晶化法制備嵌入型5Ni@S1、15Ni@S1催化劑:取上述焙燒后的2.72 g Ni/Q10粉碎后放入研缽,再緩慢滴加3.25 g四丙基氫氧化銨(質量分數25% TPAOH)并研磨30 min,使TPAOH和Ni/Q10充分混合均勻。將上述混合物轉移至水熱釜中,將水熱釜放置在200 ℃恒溫箱中反應48 h。取出產物過濾,100 ℃恒溫干燥12 h,550 ℃空氣氛圍中焙燒4 h,冷至室溫后造粒成0.850~0.425 mm(20~40目)儲藏備用。制備過程如圖1所示。
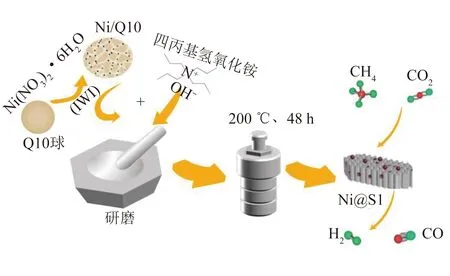
圖1 研磨-晶化制備示意Fig. 1 Schematic diagram of grinding-crystallization preparation
1.2 催化劑活性評價
催化劑活性評價采用H2還原和CH4-CO2重整反應在固定床反應裝置同時進行的方式。稱量0.2 g粒度0.850~0.425 mm(20~40目)的重整催化劑,裝入φ=8 mm的石英管反應器內并用石英棉固定。采用流速40 mL/min的N2作為保護氣,以10 ℃/min升溫速率吹掃70 min,使溫度達到700 ℃。700 ℃后,切換成H2進行還原。還原2 h后切換成反應氣(CH4/CO2/Ar=44.0/47.2/8.8,流速為40 mL/min)開始連續反應6 h,該反應為常壓過程。反應過程中每隔30 min取1次樣。出口產物采用GC-TCD在線分析,計算反應物CH4、CO2轉化率,產物H2和CO的選擇性和H2/CO,方法如下:
X(CH4)=
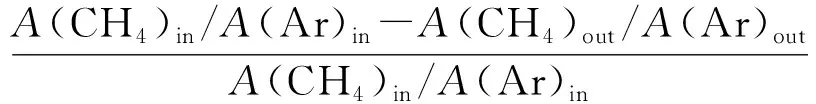
(1)
X(CO2)=
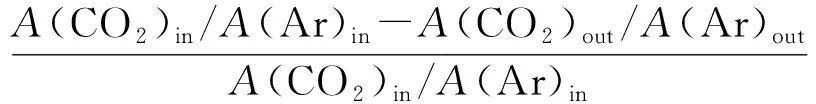
(2)
S(H2)=
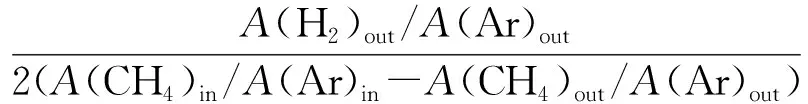
(3)
(4)
(5)
式中,A為峰面積;in和out分別代表進口和出口;X為轉化率;S為選擇性;γ為氫碳比。
1.3 催化劑表征
催化劑物相在日本理學公司Rigaku Ultima IV型X射線衍射儀上分析。輻射源CuKα,管電壓和電流分別為40 kV 和40 mA,功率1.6 kW,測量范圍5°~80°,掃描步長0.02°,掃描速度5 (°)/min。
催化劑比表面積和孔結構在美國康塔Autosorb-IQ-C全自動物理化學吸附儀上測定。樣品分析量為0.1 g,先在200 ℃對樣品進行真空處理,持續3 h,接著用-196 ℃的N2進行物理吸附。樣品比表面積和孔結構等參數用Brunauer-Emmett-Teller方程計算,而孔徑分布則由脫附等溫曲線估算得到。
催化劑原位紅外光譜表征在配有原位紅外樣品池的Nicolet Nexus型傅里葉變換紅外光譜儀上進行,將研細的催化劑粉末與KBr一起壓制成直徑13 mm的薄片進行檢測。
催化劑還原行為在日本麥奇克拜耳公司的全自動三站化學吸附儀(BELCAT-B3)進行測試,稱取0.05 g樣品,先用流速25 mL/min的Ar在50 ℃吹掃1 h,然后用10%的H2/Ar混合氣以30 mL/min流速進行還原,以10 ℃/min升溫速率從50 ℃升至800 ℃,在升溫還原過程中H2消耗量用在線TCD檢測。
催化劑酸堿性在日本麥奇克拜耳公司的全自動三站化學吸附儀(BELCAT-B3)進行測試。稱取0.05 g樣品,NH3吸附前先在300 ℃對樣品進行預處理,用流速30 mL/min的He吹掃1 h,待溫度降至100 ℃時,用5% NH3以同樣流速吹掃30 min,然后用He吹掃30 min。NH3脫附在He吹掃下進行,以10 ℃/min升溫速度升至700 ℃,NH3脫附量采用在線TCD檢測。
催化劑形貌在美國FEI公司Quanta 400 FEG型掃描電鏡(SEM)下觀察,測試電壓15 kV,測試電流為102 μA。測試前將粉末樣品進行真空干燥,噴金處理,以增加樣品的導電性。
采用美國公司FEI生產的Tecnai G2 F20 型場發射電子透射顯微鏡(TEM)進行測量,點分辨率為0.248 nm,線分辨率為0.102 nm,加速為200 kV。
催化劑X射線熒光光譜分析在美國賽默飛PERFORM′X型波長色散X射線熒光光譜儀上進行。
催化劑的X射線能譜分析在賽默飛世爾ESCALAB 250XI X射線光電子能譜熱譜儀上進行。采用MgKα輻射源,稱量0.05 g粉末樣品,在100 ℃干燥4 h后進行測量。采用XPS Peak Fit 4.1軟件對分析結果進行解析和分峰擬合。
催化劑熱性能在德國耐馳同步熱分析儀上分析。稱取樣品10 mg,用流速20 mL/min空氣吹掃,用流速10 mL/min的N2作為保護氣,以10 ℃/min升溫速度升至900 ℃,得到TG和DSC曲線。
2 結果與討論
2.1 催化劑的物相分析
圖2為載體和鎳基催化劑反應前和反應后的XRD圖譜。由圖2(a)可知,不同鎳負載量Ni/Q10催化劑在2θ=22°出現饅頭峰,為非晶態SiO2。相較于負載型Ni/Q10催化劑,不同鎳負載量嵌入型Ni@S1催化劑都有清晰的MFI五指峰拓撲結構(2θ=7°~30°),表明研磨-晶化法成功合成了嵌入型鎳基催化劑且鎳質量分數的增加(由5%到15%)不影響分子篩Silicalite-1結構的形成。當鎳質量分數相同時,由浸漬法制備的Ni/Q10的NiO特征衍射峰明顯高于研磨-晶化法制備的Ni@S1催化劑。這可能是由于Ni粒徑減小或Ni進入晶內。研磨-晶化法制備的Ni@S1催化劑部分氧化鎳物種進入了分子篩結構內部,從而使Ni粒子很好地嵌入分子篩結構中,進而使XRD能檢測到NiO峰值減小。其中NiO特征衍射峰為2θ=37.3°、43.4°、63.0°、75.6°、79.6°。分子篩的限域效果能有效減少鎳納米顆粒在高溫下的遷移,從而表現出高效的催化活性。在不同質量分數鎳的影響下,Ni@S1晶相的合成發生變化,當鎳質量分數由5%增加到15%時,NiO特征衍射峰逐漸增加。這說明過量負載不利于活性粒子的嵌入。由圖2(b)所知,高溫反應后出現元素金屬峰(Ni衍射峰在44.62°、51.92°、76.49°),表明NiO還原為Ni,Ni是DRM反應的活性中心。
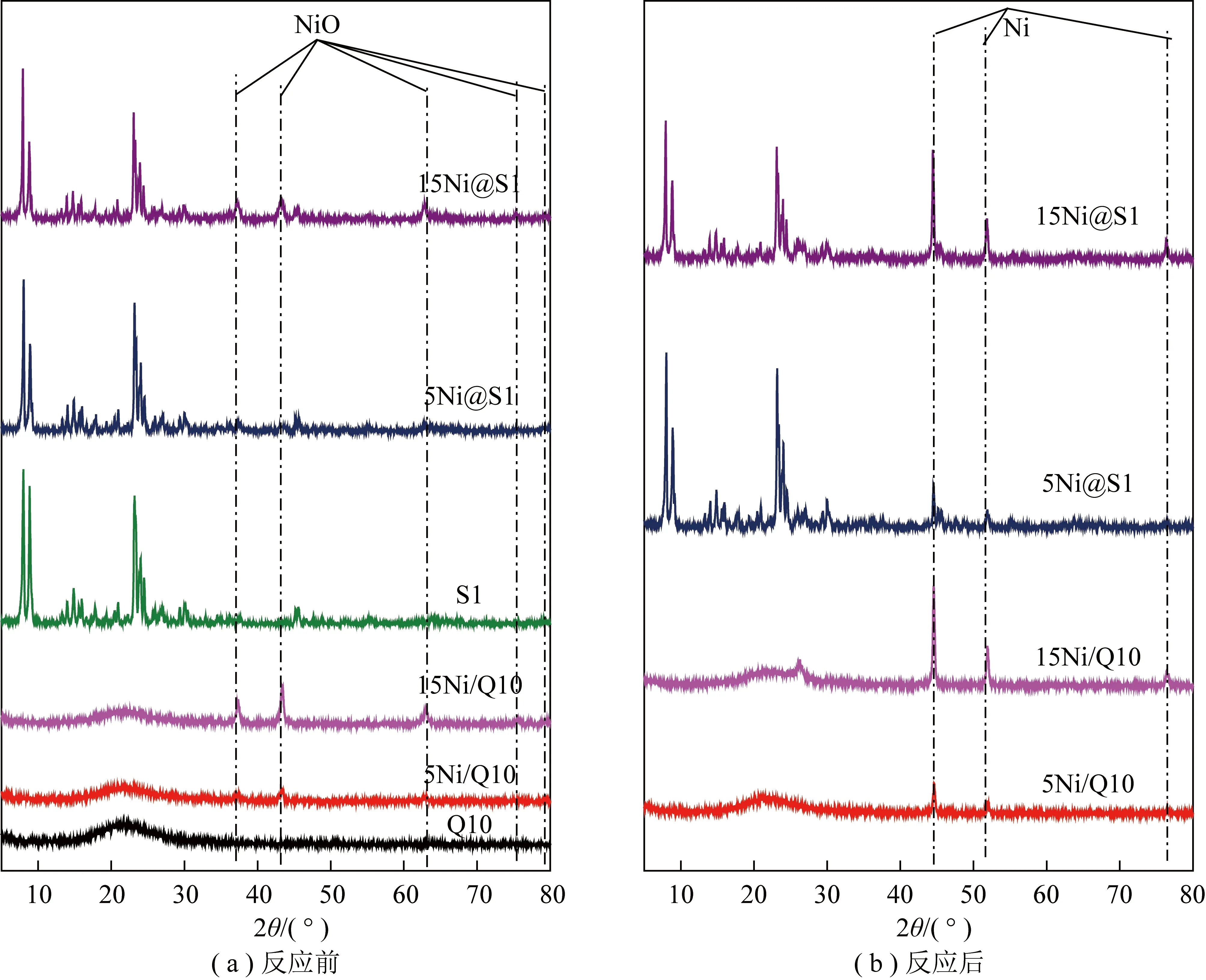
圖2 載體和不同催化劑的XRD圖Fig.2 XRD patterns for the support and different catalysts
2.2 催化劑的比表面積及孔結構分析
對載體和不同鎳基催化劑進行表面積和孔結構分析,結果如表1和圖3所示。由表1可知,采用研磨-晶化法制備的嵌入型Ni@S1催化劑與浸漬法制備的Ni/Q10催化劑相比,比表面積明顯增大,孔體積減小。比表面積增大有利于傳質的進行,從而影響后續反應過程[24]。此外,隨Ni負載量的增加(質量分數由5%到15%),比表面積進一步減小,也說明了活性金屬成功負載。將反應后不同質量分數鎳的Ni@S1催化劑與反應前對比,發現比表面積和孔體積減少,這可能是產生少量碳沉積所致。由圖3可知,相比于無約束Ni/Q10催化劑,嵌入型Ni@S1催化劑的介孔消失,形成微孔結構。微孔結構更有利于抑制小顆粒鎳在高溫下的遷移過程,起到物理屏障的作用。這說明研磨-晶化法可以將原介孔Ni/Q10有效轉變為微孔占主導地位的Ni@S1催化劑,并能將Ni活性相封裝在分子篩Silicalite-1中,這與上述XRD表征結果一致。Ni負載量改變時,嵌入型Ni@S1催化劑吸附等溫線發生變化而孔徑分布無明顯變化。

表1 載體和不同催化劑的孔結構性質
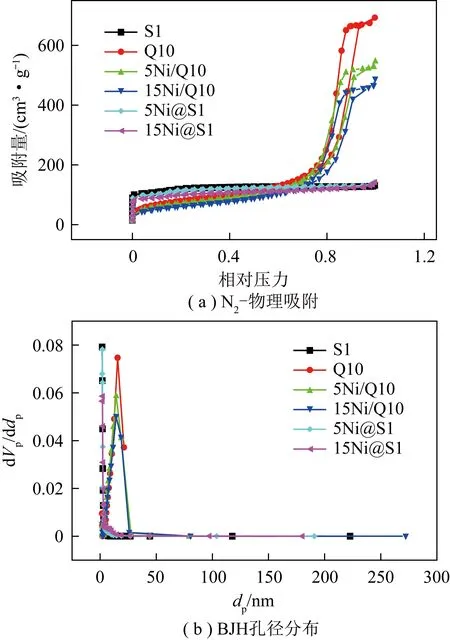
圖3 載體和反應前催化劑表面積和孔結構分析Fig.3 Surface area and pore structure plots for the support and fresh catalysts
2.3 催化劑的紅外分析
煅燒后的載體和催化劑的FI-IR圖如圖4所示。由圖4可知,所有催化劑均在3 450 cm-1觀察到較寬的O—H拉伸振動峰,這與氫鍵的O—H和自由水的羥基有關。由圖4可知其強度隨Ni負載量的增大(質量分數由5%到15%)而減小。此外,在1 640 cm-1有微弱的O—H彎曲振動峰值,這可能是樣品或KBr從空氣中吸水所致。圖4中位于1 095 cm-1的譜峰為Si—O—Si的非對稱伸縮振動吸收峰,802和470 cm-1處的譜峰分別對應于Si—O—Si 的對稱伸縮和彎曲振動。但對于嵌入型Ni@S1催化劑而言,在400~900 cm-1未觀察到Ni—O的拉伸振動,這是由于活性金屬被封裝在分子篩中,這與XRD的表征結果相一致。
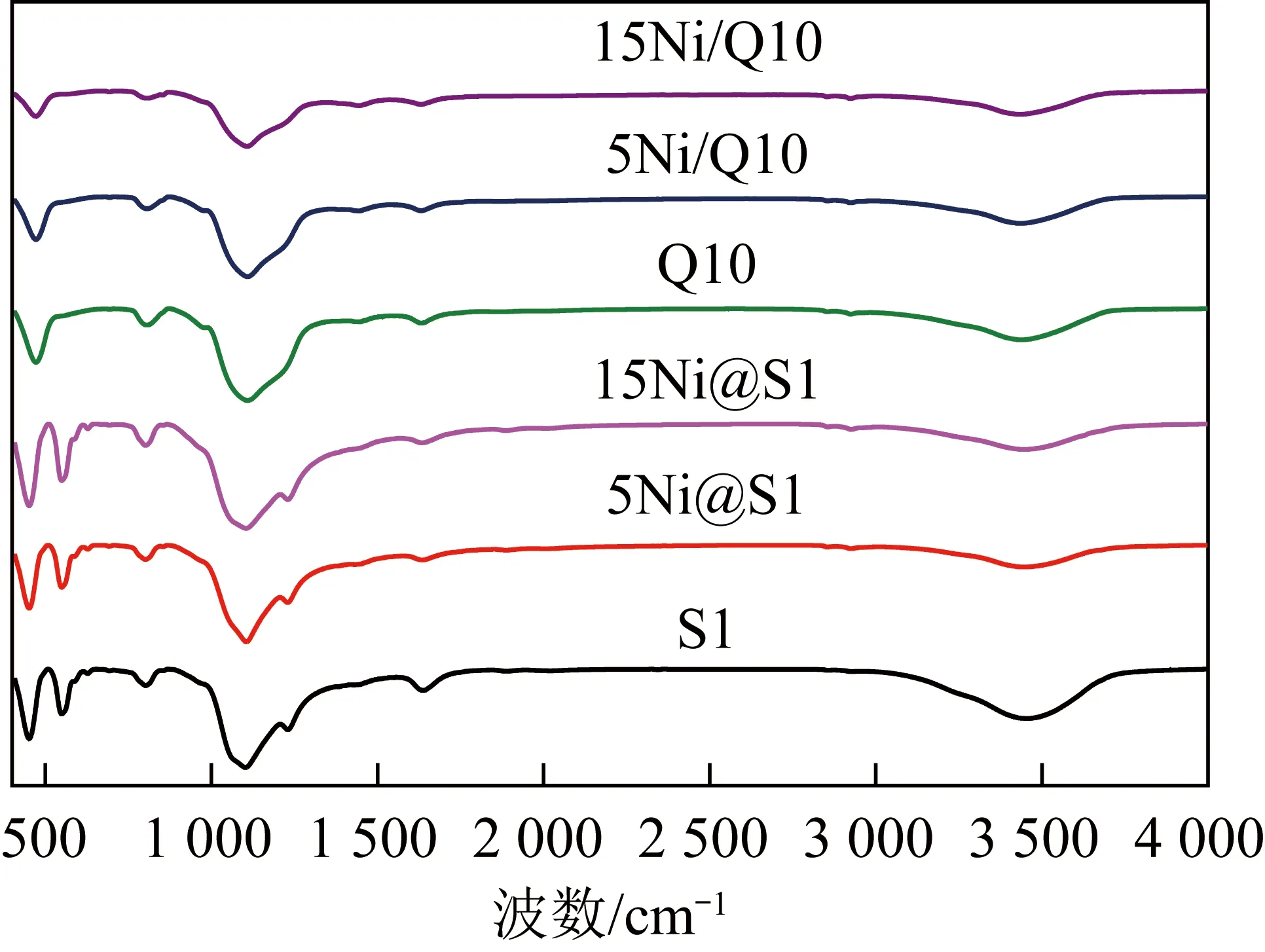
圖4 載體和反應前催化劑FI-IR譜圖Fig.4 FTIR spectra for the support and fresh catalysts
2.4 催化劑的還原性分析
采用H2-TPR研究了Ni/Q10和Ni@S1催化劑的還原行為,如圖5所示,所有催化劑上均觀察到NiO的還原峰,但還原峰的面積和位置隨催化劑結構和Ni負載量的改變而不同。當Ni質量分數為5%時,嵌入型Ni@S1催化劑與Ni/Q10催化劑相比,NiO還原峰的溫度由360 ℃右移動到425 ℃,說明活性組分與載體的相互作用增強,進一步說明封裝結構有利于抑制Ni納米顆粒的遷移,從而提高催化劑在DRM反應的活性和穩定性。當Ni質量分數為15%時,Ni@S1催化劑與Ni/Q10催化劑還原峰位置無明顯變化,可能是Ni質量分數過高導致部分Ni無法封裝在分子篩結構中而暴露在表面所致。此外,隨著質量分數從5%增加到15%,還原峰面積增加,用于反應的活性相粒子增加。
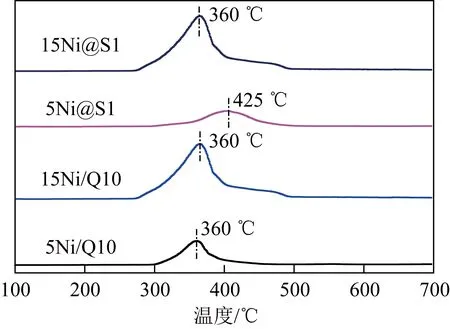
圖5 反應前催化劑的H2-TPR圖Fig.5 H2-TPR profiles of fresh catalysts
2.5 催化劑的酸性分析
載體和催化劑的NH3-TPD測試結果如圖6所示。由圖6可以看出,在115~215、215~330和345~540 ℃處的峰分別為弱酸性、中酸性和強酸性位點。Q10、S1幾乎難以發現NH3脫附峰,這是因為Q10和Silicalite-1僅具有微弱的酸性,在正常條件下不易被檢測。負載型催化劑5Ni/Q10、15Ni/Q10催化劑出現強酸性位點。除此之外,嵌入型催化劑15Ni@S1則出現了弱酸性位點,這種酸性變化可能是由于金屬氧化物與Silicalite-1分子篩孔道內硅缺陷位的羥基窩發生相互作用從而產生Lewis酸。酸性位點有助于反應物的活化,但是不利于消碳反應進行,這可能導致催化劑容易積碳失活。因此與負載型Ni/Q10催化劑相比,酸性位點的減少是嵌入型Ni@S1催化劑保持高穩定性的一個非常重要的原因。
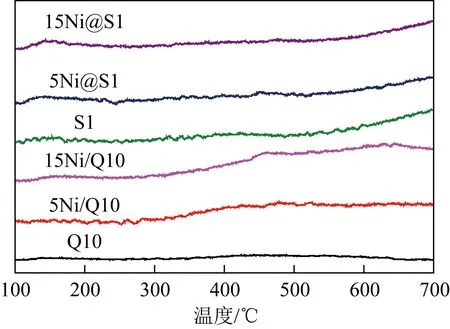
圖6 載體和反應前催化劑的NH3-TPD圖Fig.6 NH3-TPD profiles for the support and fresh catalysts
2.6 催化劑的X射線熒光光譜分析和X射線能譜分析
表2為不同催化劑的XRF總體元素含量和XPS表面元素含量結果。XRF結果表明,添加相同質量分數鎳的Ni/Q10和Ni@S1催化劑中總體Ni含量相近,表明Ni進入催化劑中。XPS結果表明Ni@S1催化劑表面Ni含量高于Ni/Q10催化劑,可能是負載型Ni/Q10的孔道大,鎳大部分負載在載體Q10孔道中。而對于嵌入型Ni@S1催化劑而言,由于微孔結構占主導地位,一部分Ni在晶體成核過程中嵌入晶體,而一部分停留在催化劑表面。圖7為不同催化劑XPS譜圖。由圖7(a)可以觀察到當Ni/Q10催化劑轉變為封裝型Ni@S1催化劑后,854.6 eV峰面積明顯減小,但出現了856.6 eV峰。一般來說,煅燒后催化劑的Ni2+2p在約854.6 eV處顯示出峰值對應于NiO。而856.6 eV處的峰值對應于Ni-O-Si物種。這表明嵌入型Ni@S1催化劑使Ni和SiO2之間的連接變得更強,這可能由于NiO被封裝在分子篩Silicalite-1中,導致各種Ni-O-Si物種產生,這與上述XRD和H2-TPR表征一致。圖7(b)顯示了2種不同催化劑氧化學狀態。O 1s峰分別為載體中的O(SiO2)(532.9 eV)、表面O(O(surf),531.8 eV)和金屬氧化物氧原子的晶格O(O(latt),530.0 eV)。顯然封裝型Ni@S1催化劑表面O含量顯著增加,表面O容易與CO2空軌道相互作用,從而通過產生各種碳酸鹽結構吸附活化CO2分子。在CH4-CO2重整反應中,表面O位點對于CO2活化至關重要。
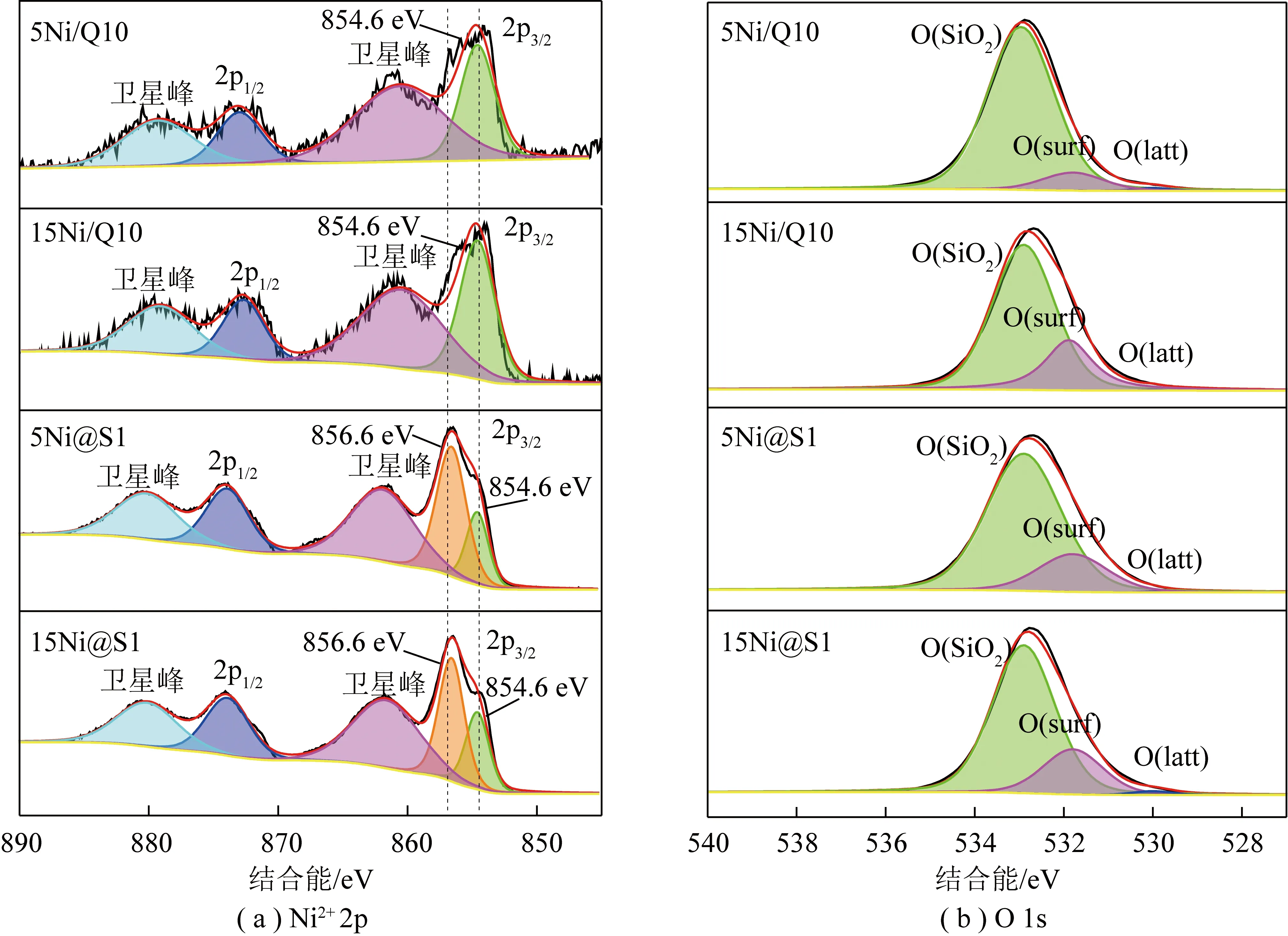
圖7 不同催化劑的XPS譜Fig.7 XPS spectra of different catalysts
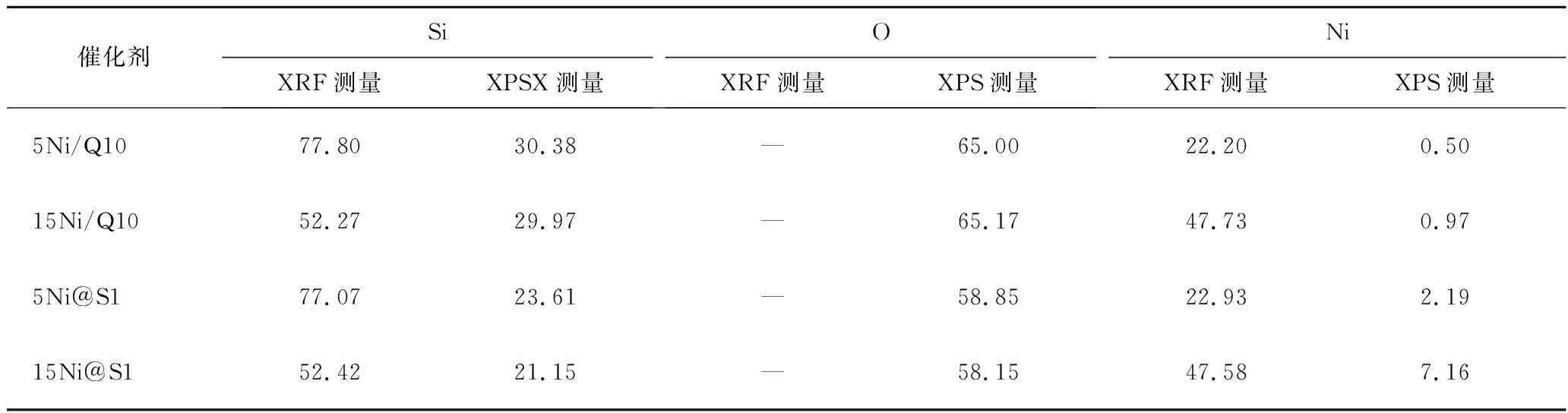
表2 催化劑元素質量分數
2.7 催化劑的形貌分析
Ni/Q10和Ni@S1的反應前SEM圖如圖8所示,由圖8可知,采用研磨-晶化法合成的S1、5Ni@S1、15Ni@S1呈現出規則的球狀,分散均勻,而Q10、5Ni/Q10、15Ni/Q10呈不規則的塊狀。此外S1、5Ni@S1、15Ni@S1 的微粒尺寸小于Q10、5Ni/Q10、15Ni/Q10。
700 ℃反應后催化劑的SEM圖如圖9所示。與反應前(圖8)對比,可以觀察到5Ni@S1,15Ni@S1反應前后形貌和顆粒大小沒有明顯變化,說明研磨-晶化法合成的催化劑具有較高的耐熱穩定性。此外在催化劑表面沒有觀察到明顯積碳,說明該鎳基嵌入型結構催化劑還具有抗積碳的特性。對比5Ni/Q10,15Ni/Q10出現了塊狀聚集和表面積碳。
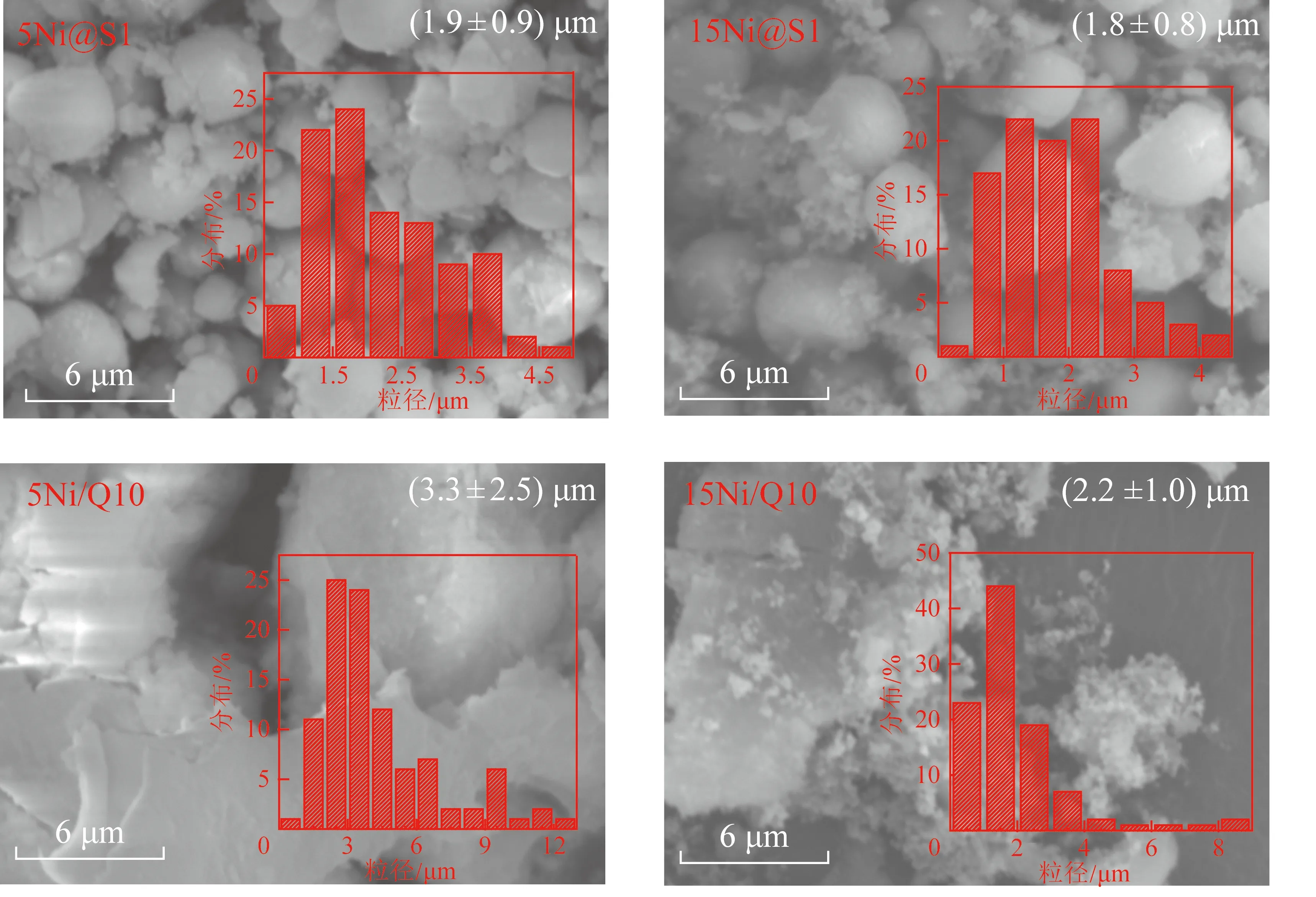
圖9 反應后催化劑SEM圖Fig.9 SEM images for the spent catalysts
為進一步明確嵌入型結構催化劑中Ni金屬的嵌入狀態,對700 ℃ H2還原后的5Ni@S1催化劑進行TEM表征,如圖10所示。圖10顯示了Ni納米顆粒嵌入分子篩Silicalite-1中,說明Ni/Q10作為硅源在參與形成分子篩Silicalite-1過程中發生了原位轉化。這種催化劑結構能夠抑制鎳納米顆粒在高溫下的遷移以及積碳的形成,從而影響DRM反應。
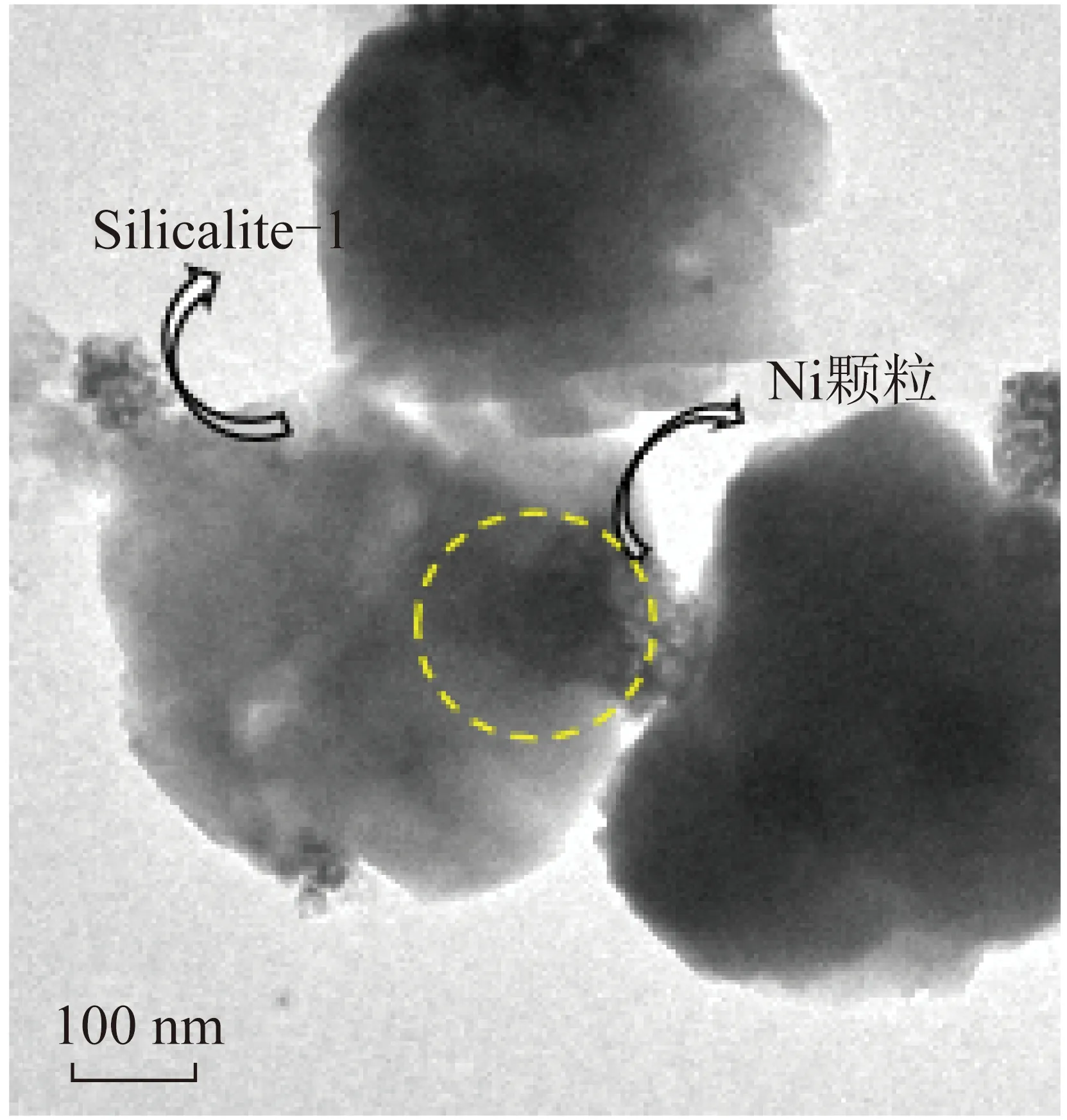
圖10 還原后5Ni@S1催化劑TEM圖Fig.10 TEM images of reduced 5Ni@S1 catalyst
2.8 Ni含量對催化活性影響
對不同催化劑進行DRM反應測試,結果如表3和圖11所示。可知在相同Ni負載量情況下,Ni@S1催化劑比Ni/Q10催化劑具有更加優異的活性和穩定性。在700 ℃進行DRM反應6 h后,Ni@S1轉化率沒有明顯變化,穩定性明顯高于Ni/Q10催化劑,積碳量也明顯減少,這得益于Ni@S1催化劑的物理和化學雙重限域效益,在高溫下抑制鎳粒子遷移,使催化劑保持較高的穩定性。反應6 h后,5Ni@S1和5Ni/Q10催化劑的CH4最終轉化率分別為72.82%和67.24%,與初始轉化率相比分別降低了1.05 和7.99個百分點;CO2最終轉化率分別為79.06% 和76.69%,與初始轉化率相比分別降低了1.16和4.54個百分點。15Ni@S1活性的降低可能是由于鎳的質量分數過高導致部分鎳暴露在分子篩表面團聚引起活性降低。此外,由于逆水煤氣變換反應,導致CO2轉化率高于CH4轉化率,因此產物中H2/CO小于1。反應6 h后,15Ni@S1催化劑的H2/CO(0.94)大于15Ni@Q10催化劑的H2/CO(0.88),這可能是嵌入型結構能一定程度抑制逆水煤氣變換反應的結果。
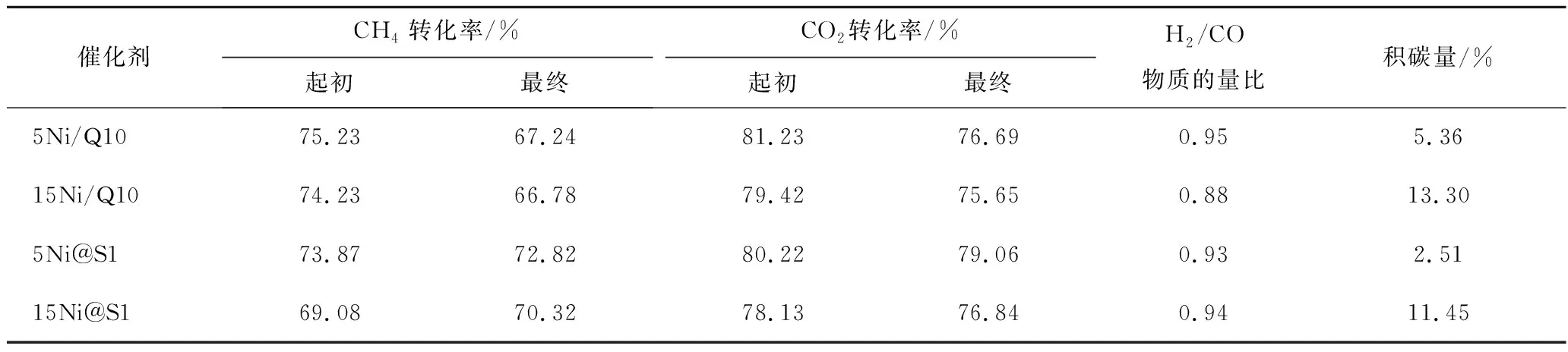
表3 CH4-CO2 重整反應活性
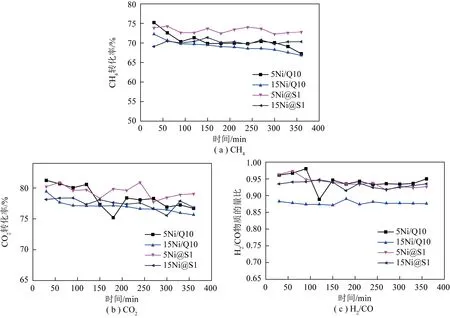
圖11 不同催化劑上催化活性隨在線反應時間變化Fig.11 Time-conversion diagram of different catalysts
2.9 反應后催化劑積碳分析
圖12為不同催化劑在反應溫度700 ℃下DRM反應6 h后的積碳燃燒曲線。由圖12可知,所有催化劑在0~100 ℃均產生失重臺階,這是由于反應后催化劑中未完全干燥殘留水分蒸發導致。在400~600 ℃產生了一個明顯失重平臺,對應DSC曲線明顯的放熱峰,歸應于積碳的燃燒。研究發現,當Ni負載量相同時,Ni@S1催化劑的積碳比Ni/Q10催化劑少。其中,5Ni@S1催化劑的積碳量僅為5Ni/Q10催化劑的46.83%。嵌入型Ni@S1催化劑Ni質量分數由5%增大到15%的過程中,積碳量增加,這與DRM反應活性和穩定性規律一致。15Ni@S1仍有大量積碳,可能是由于負載量過高導致鎳活性物種無法全部嵌入分子篩Silicalite-1中,暴露在表面導致CH4在催化劑表面快速裂解,積碳反應速率大于消碳反應速率從而導致積碳量增加。
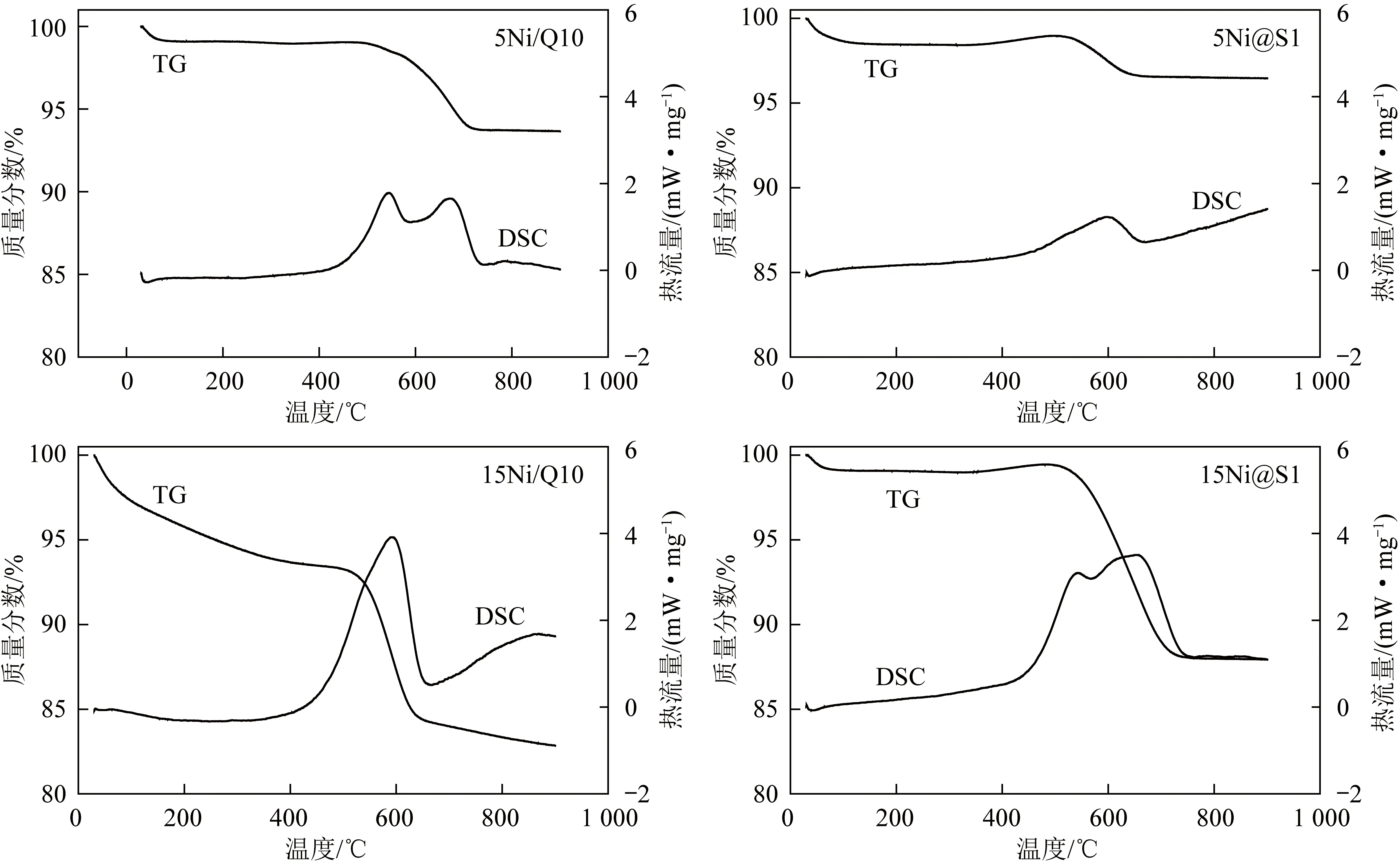
圖12 700 ℃反應后6 h催化劑TG-DSC圖Fig.12 TG-DSC diagram of spent catalysts after reaction at 700 ℃ for 6 h
3 結 論
1)當Ni的負載量相同時,與Ni/Q10催化劑相比,Ni@S1催化劑具有更好的催化活性、抗燒結性和抗積碳性。Ni@S1催化劑中活性金屬顆粒尺寸更小,同時被固定在分子篩內部,分子篩與金屬之間形成強相互作用。嵌入型結構還能夠減少催化劑積碳,5% Ni@S1催化劑的積碳量僅為5% Ni/Q10催化劑的46.83%。
2)催化劑用于700 ℃下DMR反應6 h后,CH4在5Ni@S1和5Ni/Q10催化劑的最終轉化率分別為72.82%和67.24%,與初始轉化率相比分別降低了1.05 和7.99個百分點;CO2最終轉化率分別為79.06% 和76.69%,與初始轉化率相比分別降低了1.16和4.54個百分點。相比于負載型催化劑,嵌入型催化劑表現出更好的活性和穩定性。當Ni負載量為15%,部分Ni會殘留在分子篩表面并在高溫反應中發生聚集,影響催化活性和穩定性。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
