可磁分離回收多孔CoFe2O4的制備及其催化過一硫酸鹽降解亞甲基藍溶液的性能
2022-06-15 05:54:06向小倩廖小剛胡學步
材料工程 2022年6期
夏 強,向小倩,廖小剛,鄭 林,李 綱,胡學步
(重慶理工大學 化學化工學院,重慶400054)
印染行業排放的偶氮類染料廢水具有色度深、有機質含量高、可生化性差、毒性強和排放量大等特點[1],屬于典型的難降解工業廢水,對生態環境和人類健康具有潛在威脅。因此,如何從源頭上實現對染料廢水的高效處理以確保其穩定達標排放是目前生態環保領域所面臨的一項重大課題。高級氧化技術(advanced oxidation processes, AOPs)因具有對有機污染物處理速度快、降解效率高和適用范圍廣等優點,而在水處理領域被廣泛采用[2-3]。該技術得以成功運行的關鍵在于高活性催化材料的獲得。催化劑按照使用形式,可分為均相和非均相兩類。較之非均相催化劑,均相催化劑盡管因與反應物分子接觸更充分而通常顯示出較為優異的催化活性,但卻也存在著在使用后難于回收而易造成水體被二次污染的缺點。因此,有關AOPs中可回收的非均相催化劑的開發及其在水處理應用方面的研究越來越受到科研工作者的重視。
鑒于Fe2O3和Co3O4材料均可活化PMS,并考慮到鐵氧化物通常表現出的良好磁性能,本工作提出設計構建具有磁分離回收特性的鐵鈷二元復合氧化物材料作為非均相催化劑。材料的制備采用草酸鹽熱分解兩步法完成,即首先通過草酸鹽沉淀法制得鐵鈷草酸鹽前驅體,隨后熱分解該前驅體獲得產物。該方法的優點在于,草酸鹽熱分解時可原位產生孔洞,從而獲得多孔鐵鈷二元復合氧化物,有助于進一步提高材料的催化性能。選擇典型的陽離子型染料亞甲基藍(MB)作為降解模型,以評價該復合材料催化活化PMS降解處理模擬印染廢水的性能。采用單因素實驗研究工藝參數對催化劑活化PMS性能的影響,從而獲得優化的MB溶液降解工藝條件。此外,采用猝滅實驗和EPR技術對反應過程中產生的活性物種進行鑒別,進而提出鐵鈷二元復合氧化物材料活化PMS降解MB溶液的反應機理。
1 實驗部分
1.1 試劑
乙二醇、無水乙醇、七水合硫酸鈷、甲醇(MeOH)、叔丁醇(TBA)、L-組氨酸(L-Histidine)、氯化硝基四氮唑藍(NBT)及亞甲基藍(MB),購于上海泰坦科技有限公司;二水合草酸、七水合硫酸亞鐵、氯化鈉、草酸鈉和磷酸鈉,購于成都科隆化學品有限公司。實驗所用試劑均為分析純。實驗用水為自制去離子水,在GWA-UN2超純水器(北京普析通用儀器有限公司)上制得。
1.2 材料的制備
按照一定的Fe/Co摩爾比(1∶0,2∶1和0∶1)稱取FeSO4·7H2O與CoSO4·7H2O共12 mmol溶于160 mL乙二醇中,記為溶液A;稱取12 mmol H2C2O4·2H2O溶入80 mL去離子水中,記為溶液B。在強烈攪拌下,將溶液A快速倒入溶液B中,并繼續攪拌20 min。隨后,將該混合液轉移至容積為500 mL帶有聚四氟乙烯內襯的水熱釜中,密封后置于120 ℃的烘箱中水熱處理16 h。反應結束后,離心收集沉淀,并依次用去離子水和無水乙醇洗滌數次,于50 ℃干燥,即得草酸鹽前驅體。將草酸鹽前驅體置于馬弗爐中煅燒獲得最終產物,煅燒溫度為400 ℃,時間為2 h,升溫速率為2 ℃·min-1。為方便記錄,將前述三種不同Fe/Co摩爾比金屬鹽投加量下得到的產物依次命名為S1,S2和S3。
1.3 分析表征
樣品的晶相結構和相純度測試在XRD-7000型X射線衍射儀上完成,Cu靶,Kα輻射,加速電壓和電流分別為40 kV和40 mA,2θ掃描范圍為10°~80°;采用Quanta 600F型掃描電鏡對樣品的形貌進行觀察;N2吸附-脫附曲線在Micrometritics ASAP2020氮氣吸附儀上完成,工作溫度為-195.8 ℃;磁性能測試在PPMS-9型綜合物性測試系統上完成;XPS測試在Kratos XSAM-800型X射線光電子能譜儀上完成,Al靶,Kα射線作為發射源;自由基捕獲實驗在JES FA200型電子順磁共振波譜儀(EPR)上完成,采用5,5-二甲基-1-吡咯啉-N-氧化物(DMPO)或4-氧-2,2,6,6-四甲基哌啶(TEMP)作為自由基捕獲劑;MB溶液濃度通過紫外可見分光光度計(UV-2550)測定,波長范圍為500~700 nm。
1.4 催化性能評價
選擇典型的陽離子型染料亞甲基藍(MB)作為降解模型,以評價不同催化劑活化PMS降解有機污染物的性能。具體實驗如下:在室溫(16 ℃)下,稱取一定質量的催化劑加入到一定濃度、體積為500 mL的MB溶液中,磁力攪拌30 min,以達到吸附飽和。隨后,向上述混合液中加入一定量的PMS并開始計時,每隔10 min取樣并測量MB溶液的即時吸光度值。每次取樣量約4 mL,所取樣品經0.22 μm尼龍濾膜過濾后,與150 μL濃度3 mol·L-1的NaNO2溶液混合,以猝滅反應。MB溶液的降解率η為:
(1)
式中:C0為達到吸附平衡后MB溶液的濃度,mg·L-1;Ct為反應t時刻下MB溶液的即時濃度,mg·L-1。
2 結果與討論
2.1 表征分析結果
2.1.1 XRD分析
圖1為樣品的XRD譜圖。可以看出,S1,S2和S3的特征衍射峰分別與標準卡片JCPDS#33-0664,JCPDS#03-0864 和JCPDS#42-1467 相吻合,證實三種樣品的物相組成分別為Fe2O3,CoFe2O4和Co3O4。此外,觀察到Co3O4的特征衍射峰最為尖銳,表明其生長最為完善;Fe2O3的特征衍射峰強度居中,表明其結晶程度要弱于Co3O4;而CoFe2O4的晶化程度最低,表明Fe元素的引入抑制了Co3O4的生長。
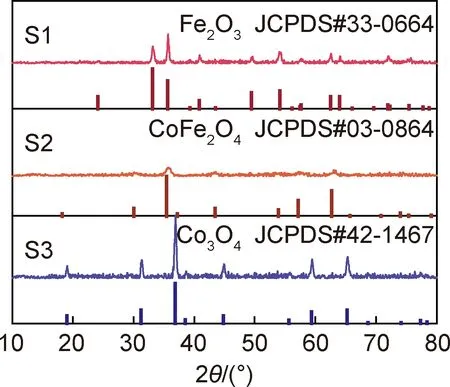
圖1 樣品的XRD譜圖
2.1.2 SEM分析
圖2為樣品的掃描電鏡圖片。從整體上看,三種材料均呈現出微米纖維結構。其中Fe2O3的均勻性最好,纖維也更加細長,單根纖維的直徑約 150~200 nm,長度約5~10 μm;較之Fe2O3樣品,單根CoFe2O4纖維的直徑略有增加,介于200~500 nm,長度則未見明顯變化,仍處于微米級尺度,且樣品整體的均勻性仍保持較好;而Co3O4樣品的形貌變得不甚規整,均勻性較差,部分區域還出現輕微團聚現象,纖維的長度仍處于微米級,但整體的長徑比明顯變小,絕大多數的單根纖維變得更為粗壯,尺寸從400 nm到1.5 μm不等。此外,觀察到三種樣品的表面均較粗糙,這是由于草酸鹽前驅體在熱處理過程中釋放出易揮發物質如H2O和CO2等所致。

圖2 樣品的SEM圖 (a)Fe2O3;(b)CoFe2O4;(c)Co3O4
2.1.3 比表面積與孔結構分析
采用N2吸附-脫附測試對樣品的比表面積和孔結構進行分析,結果示于圖3。由圖3(a)可知,三種樣品的吸-脫附曲線形狀相似,為典型的第Ⅳ類Langmiur吸附-脫附等溫線。吸附支和脫附支不重合,這是由毛細管的凝聚作用引起的,證實經由草酸鹽熱分解法所得產物具有介孔構造特征。吸附支和脫附支間的遲滯環為H3型,表明樣品的孔道為狹縫狀。圖3(b)為三種樣品對應的孔徑分布曲線。可以觀察到樣品的孔徑均分布較窄,且主要集中在介孔區域。
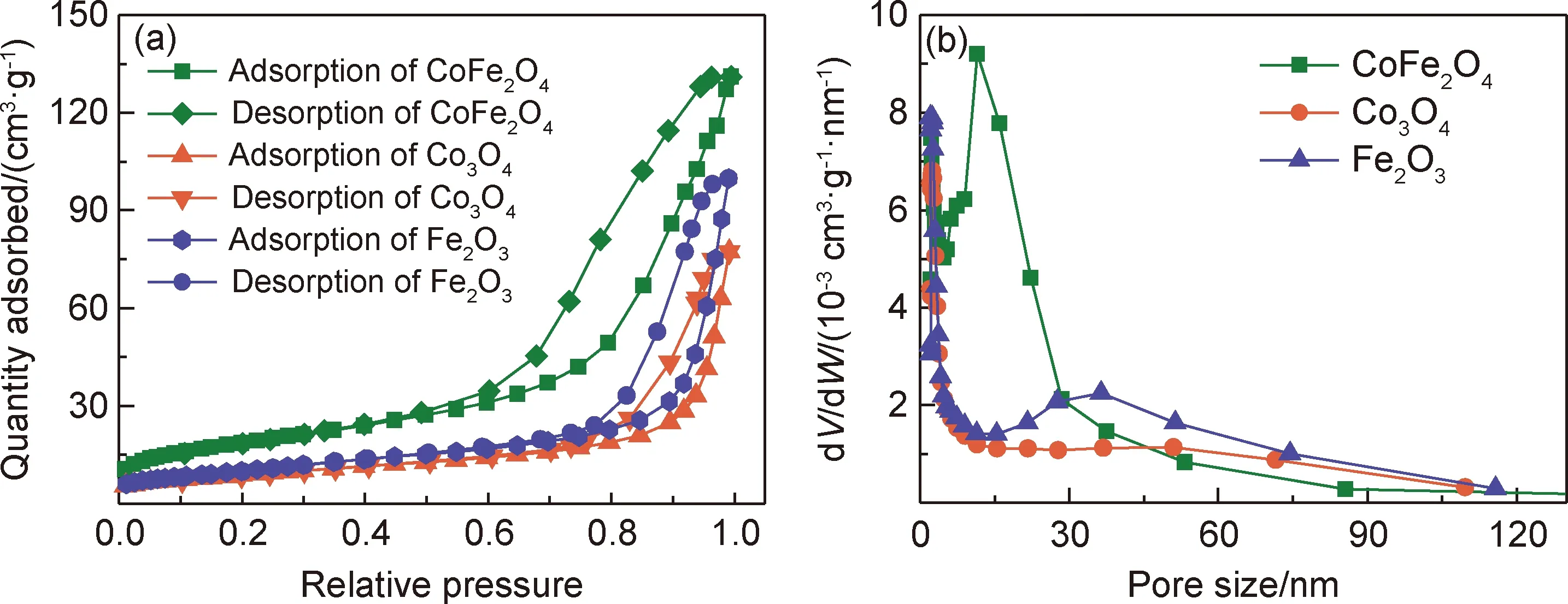
圖3 樣品的N2吸附-脫附(a)和孔徑分布(b)曲線
根據BET公式和BJH算法獲得的樣品比表面積和孔結構參數列于表1。其中CoFe2O4樣品具有最高的比表面積和最大孔容。
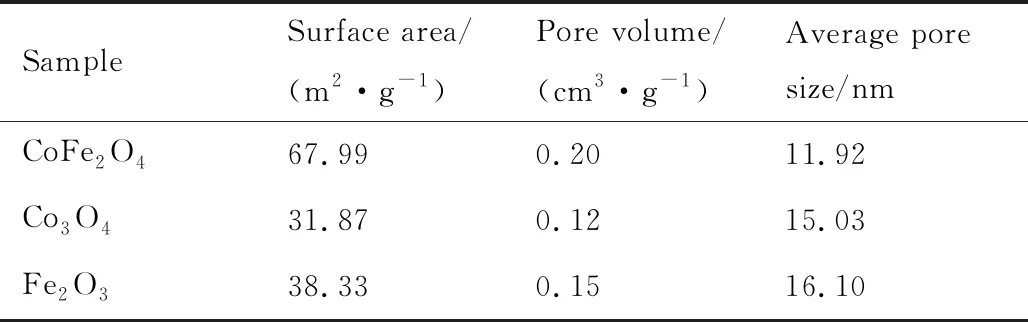
表1 樣品的孔結構參數
2.1.4 磁性能分析
圖4是在外部磁場強度為-1.59×106~1.59×106A·m-1、室溫條件下測得的三種樣品的磁滯回線。可知,CoFe2O4,Fe2O3和Co3O4的飽和磁化強度M依次為300,177.5,5 A·g-1,即CoFe2O4具有最為優良的鐵磁性,這為其作為催化劑在完成反應后借助外加磁場進行快速回收提供了便利。
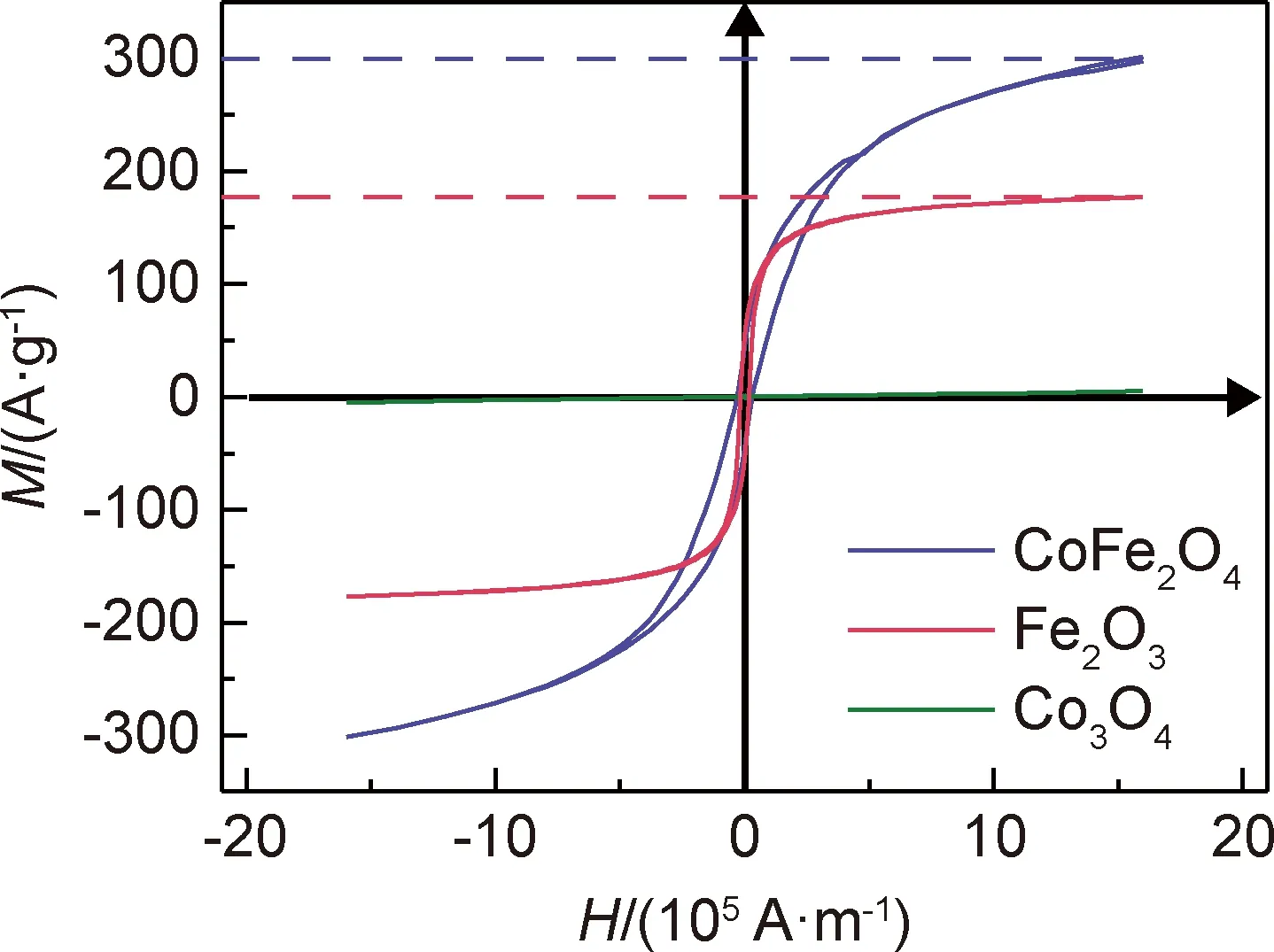
圖4 樣品的磁滯回線
2.2 MB溶液降解實驗結果
2.2.1 催化活性對比
選擇MB作為降解模型,以評價催化材料催化活化PMS降解模擬印染廢水的性能。實驗條件:催化劑添加量0.05 g,PMS用量3 mL(0.1 mol·L-1),MB溶液濃度10 mg·L-1,MB溶液體積500 mL。圖5對比了三種樣品催化PMS降解MB溶液的性能。當反應體系中僅添加PMS或CoFe2O4時,MB的去除率分別為8.75%和2.52%,少量的MB降解可分別歸因于可見光活化PMS引起的化學降解和CoFe2O4對其的物理吸附作用。而當PMS和催化劑Co3O4,CoFe2O4或Fe2O3共同存在時,MB的降解率分別為42.08%,79.31%和20.53%,按照一級反應動力學擬合得到不同催化劑存在時MB溶液的降解反應速率常數k分別為0.0104,0.0303 min-1和0.0038 min-1,是僅有PMS存在時相應速率常數(0.0020 min-1)的5.2倍、15.15倍和1.9倍,說明三種催化材料對PMS具有不同程度的活化作用。
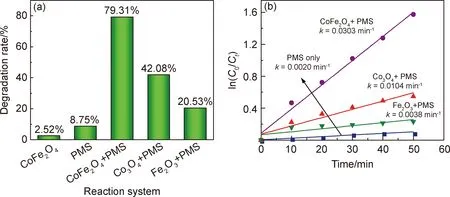
圖5 不同催化劑/PMS體系下MB溶液的降解效果對比
與單一過渡金屬氧化物Co3O4或Fe2O3相比,二元復合氧化物CoFe2O4對PMS的催化活化效果更加明顯。結合前文的材料表征結果,分析CoFe2O4具有更為優異的催化活化PMS性能的原因:CoFe2O4在三種材料中具有最高的比表面積和最大孔容,相同質量的催化劑添加量下其能夠提供更多的反應活性位點;在PMS活化過程中,鐵離子與鈷離子之間存在著一定的協同效應[5,17]。
2.2.2 不同工藝參數下CoFe2O4/PMS體系對MB溶液的降解效果
鑒于相同實驗條件下CoFe2O4/PMS體系對MB溶液的降解效果最佳,因此以CoFe2O4為研究對象,采用單因素實驗方法,系統考查相關工藝參數(催化劑添加量、PMS用量、MB溶液濃度)對其催化活化PMS降解MB溶液效能的影響,如圖6所示。
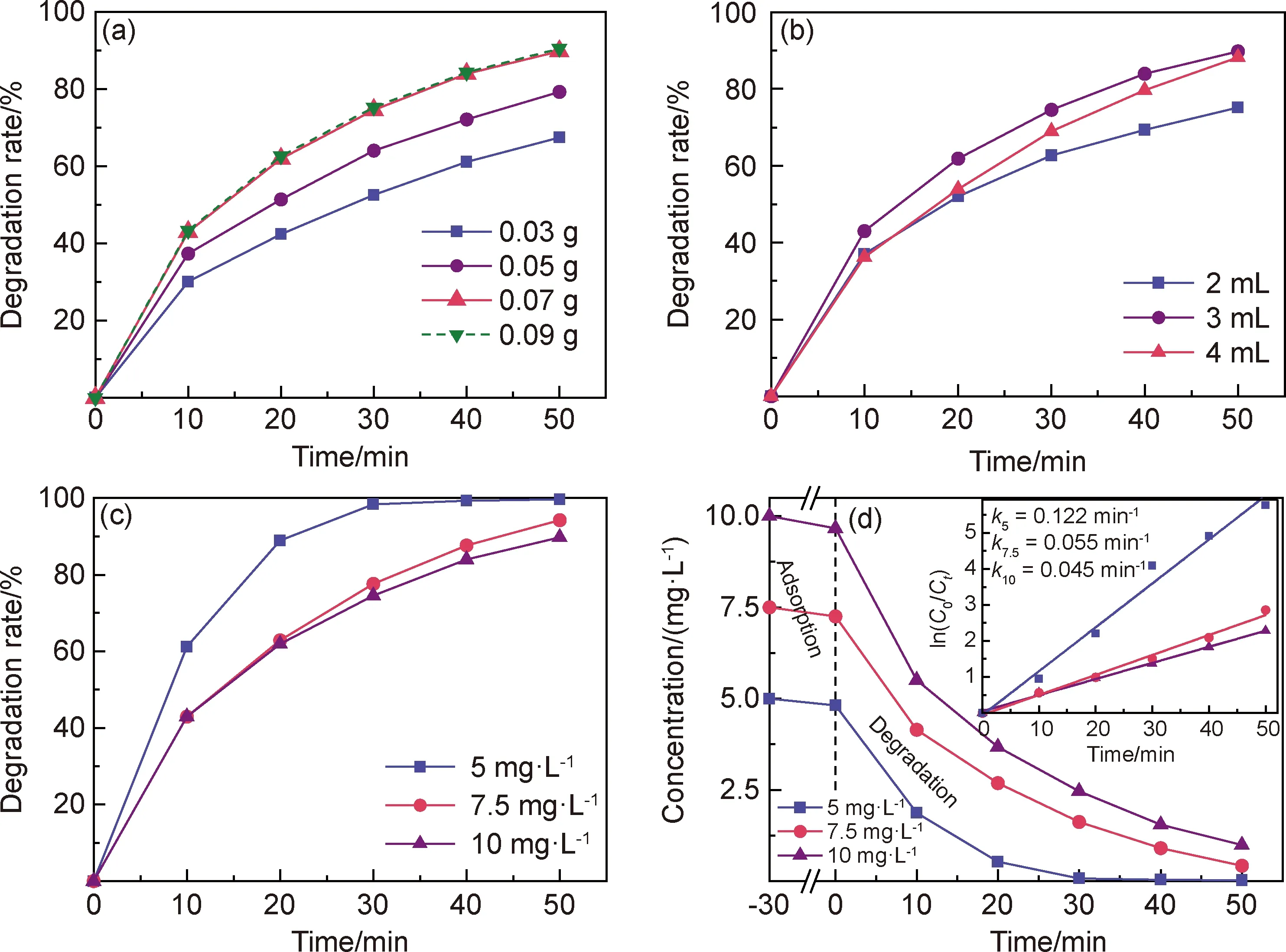
圖6 不同工藝參數下CoFe2O4/PMS體系對MB溶液的降解效果
圖6(a)為CoFe2O4添加量對催化PMS降解MB溶液性能的影響曲線。實驗時PMS用量為3 mL,MB溶液初始濃度為10 mg·L-1。可知,當催化劑添加量從0.03 g遞增至0.07 g時,MB溶液的降解率與降解速率也隨之增加。催化降解反應進行50 min后,三種催化劑添加量條件下所對應的MB溶液降解率分別為67.45%,73.31%和89.77%。這是因為,單位體積內催化劑添加量的增加可以為PMS的活化提供更多的反應活性位點,進而提升催化氧化體系對MB溶液的降解能力。但當催化劑添加量繼續增加至0.09 g時,CoFe2O4/PMS體系降解MB溶液的效果并沒有得到提升,表明在本實驗反應條件下0.07 g的催化劑添加量已達到飽和,此時催化劑所提供的反應活性位點數已足夠用于PMS的活化,且不再作為MB被氧化降解的限制性因素。
圖6(b)為PMS用量對MB溶液降解效果的影響曲線。實驗中CoFe2O4添加量為0.07 g,MB溶液初始濃度為10 mg·L-1。可知,隨著PMS用量(2,3,4 mL)的增加,反應進行50 min后MB溶液的最終降解率分別為75.14%,89.77%和88.19%,即降解效果整體上表現出先升后降的趨勢,最佳PMS用量為3 mL。體系中PMS用量過量導致MB溶液降解效果下降的原因可能為:經由催化產生的活性中間體與PMS或活性中間體相互之間發生自猝反應,造成反應體系氧化能力的下降[18-20],相關化學反應如式(2)~(4)所示。
(2)
(3)
(4)
圖6(c)為不同MB溶液初始濃度對其降解效果的影響曲線。實驗時固定CoFe2O4添加量為0.07 g,PMS用量為3 mL。可見,當MB溶液起始濃度為10 mg·L-1時,降解率為89.77%;當MB溶液起始濃度為5 mg·L-1時,降解率提升至99.68%,即隨MB溶液起始濃度的降低,MB溶液被催化降解的去除率有所提升。圖6(d)給出了不同初始濃度的MB溶液發生催化降解反應的動力學曲線。根據曲線計算得到初始濃度分別為5,7.5 mg·L-1和10 mg·L-1的MB溶液降解反應動力學速率常數依次為0.122,0.055 min-1和0.045 min-1。上述實驗數據表明,CoFe2O4/PMS體系對低濃度的MB溶液無論是在降解率還是反應速率上,均表現出更為優異的降解去除效果。這是因為,PMS被催化劑活化產生活性中間體的過程并不受MB濃度影響,但隨著MB溶液初始濃度的降低,MB分子可以有更多的機會與活性中間體發生反應,因此表現出更好的降解效果。需要指出,CoFe2O4/PMS體系對5 mg·L-1的MB溶液降解率幾乎達到100%,說明該高級氧化體系具備完全去除模擬印染廢水的能力。
2.2.3 陰離子對CoFe2O4/PMS體系降解MB溶液的影響
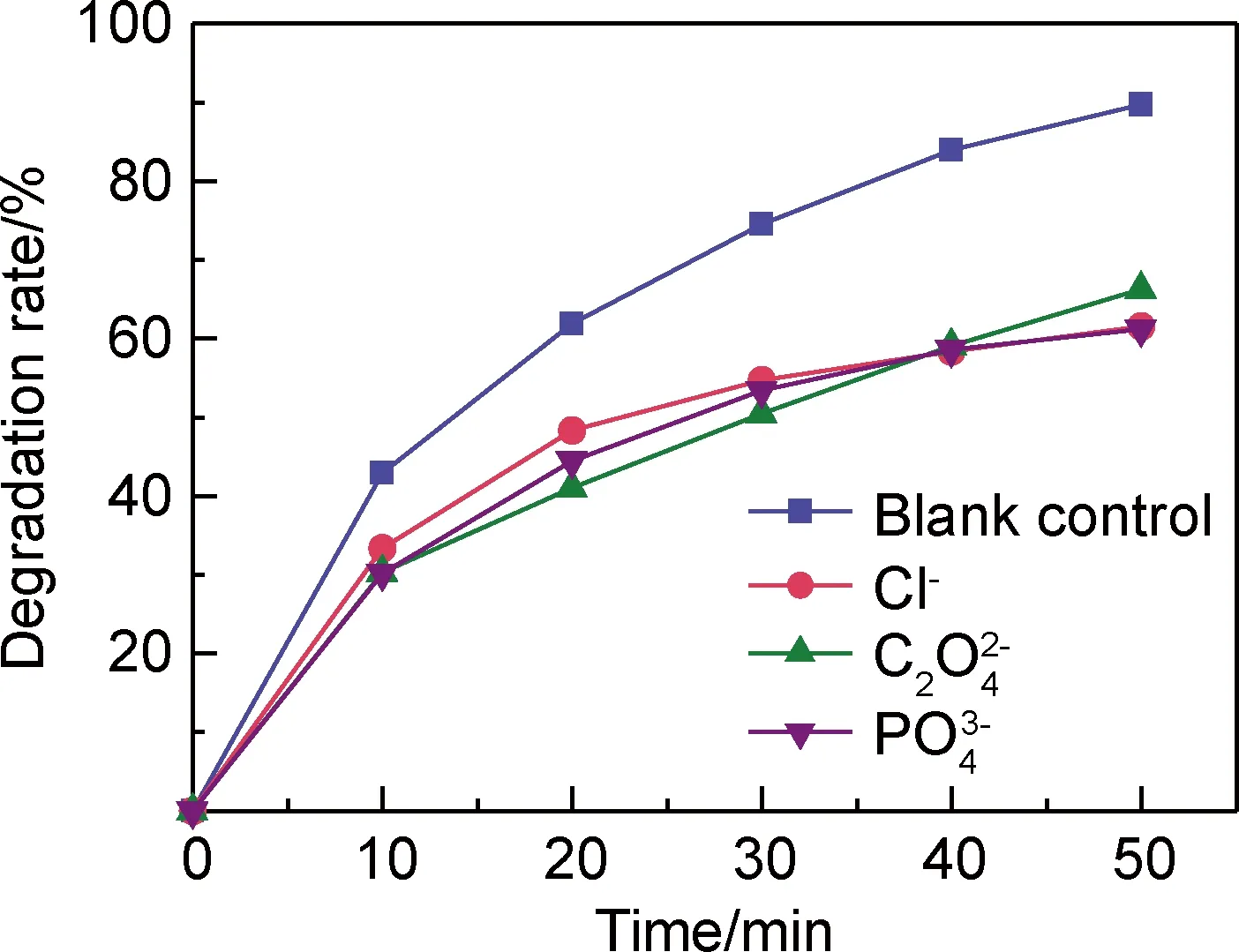
圖7 陰離子對MB溶液降解效果的影響
(5)
(6)
(7)
2.3 CoFe2O4的重復使用性能
通過施加外加磁場將使用過的CoFe2O4催化劑進行回收,用去離子水充分洗滌并干燥后,于350 ℃下煅燒2 h進行再生,以測試其作為催化劑循環用于活化PMS降解MB溶液的可行性。圖8為CoFe2O4作為催化劑循環使用3次的性能結果。曲線顯示,在1,2,3次循環后CoFe2O4/PMS體系對MB溶液的降解效率分別為89.77%,85.31%和76.67%,對應的反應速率常數依次為0.045,0.037 min-1和0.029 min-1。盡管隨循環使用次數的增加,CoFe2O4催化活化PMS降解MB溶液的效能呈現出一定的衰減趨勢,但其具有的可通過外加磁場從溶液中快速分離回收的優勢,為其在實際印染廢水處理中的應用提供了可能。

圖8 催化劑CoFe2O4處理亞甲基藍溶液的重復使用性能
為分析催化反應過程中CoFe2O4材料表面狀態的變化,對反應前后的催化劑進行XPS測試,結果如圖9所示。由圖9(a)可知,使用前的CoFe2O4材料表面檢測到Co,Fe,O及C四種元素,其中284.8 eV處出現的C1s峰信號來源于儀器自身的污染碳;催化反應后,CoFe2O4材料其表面仍檢測到Co,Fe,O及C四種元素,但C1s峰信號顯著增強,這應該與催化降解反應過程中吸附在催化劑表面的MB及其降解產物中的碳相關。圖9(b)是O1s的高分辨XPS譜圖。可見,反應前后CoFe2O4材料中的O1s均可被擬合為3個小峰,按結合能由低到高依次歸屬為晶格氧、化學吸附氧和物理吸附氧。但反應之后催化劑表面的吸附氧含量明顯增多,進一步證實反應后催化劑的表面存在難以去除的吸附物。圖9(c)為Fe2p的高分辨XPS譜圖。該譜可被擬合為2個歸屬于Fe(Ⅱ)的峰(即724 eV處的2p1/2峰和710 eV處的2p3/2峰)、2個Fe(Ⅲ)的峰(即726 eV處的2p1/2峰和712 eV處的2p3/2峰)以及2個衛星峰,這證實CoFe2O4材料中的Fe同時以Fe(Ⅱ)和Fe(Ⅲ)兩種價態共存。進一步觀察發現,反應前后催化劑Fe2p譜的形狀及Fe(Ⅱ)/Fe(Ⅲ)間的相對原子比均未發生大的變化。圖9(d)為Co2p的高分辨XPS譜圖。該譜可被擬合為歸屬于Co(Ⅱ)的峰(即796 eV處的2p1/2峰和781 eV處的2p3/2峰)、Co(Ⅲ)的峰(即795 eV處的2p1/2峰和779 eV處的2p3/2峰)以及2個衛星峰,表明CoFe2O4材料中的Co同時以Co(Ⅱ)和Co(Ⅲ)兩種價態共存。對比反應前后的譜圖可發現,CoFe2O4材料中Co(Ⅱ)/Co(Ⅲ)間的相對原子比變化較大,反應后Co(Ⅲ)的含量大幅上升。文獻報道[23-25],PMS被活化是金屬離子與PMS分子間發生單電子轉移的結果。基于XPS分析,不難發現CoFe2O4催化劑活化PMS時鈷離子的作用更大。至于CoFe2O4在循環使用后其催化性能有所下降,原因可能:部分有機物以及降解中間產物附著在催化劑表面難以去除,占據反應活性位點;循環使用后催化劑中過渡金屬離子價態分布發生變化;催化劑表面的活性位點在多次使用過程中可能存在一定的流失。
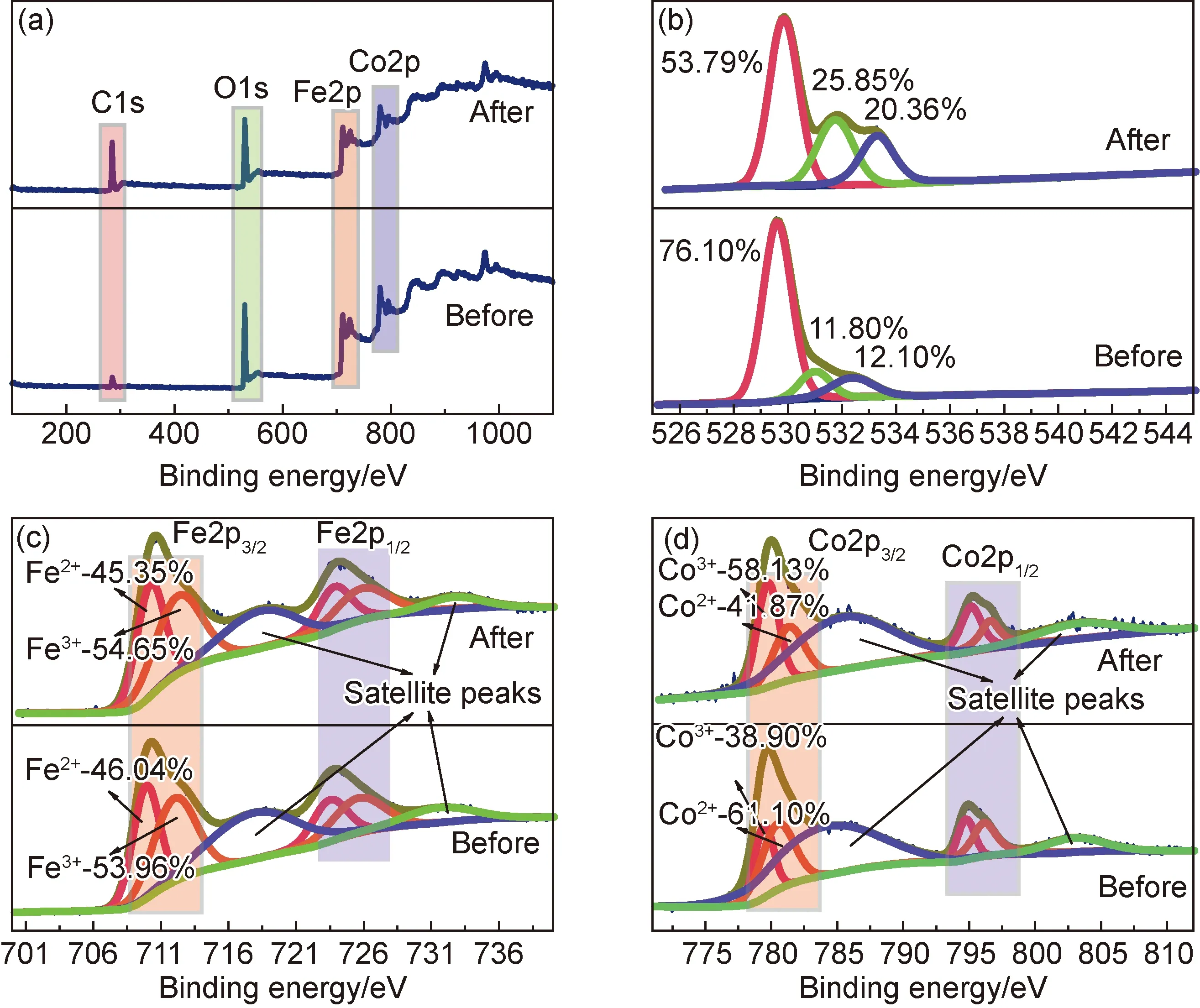
圖9 反應前后CoFe2O4催化劑的XPS分析
2.4 活性中間體鑒定與PMS活化機理分析
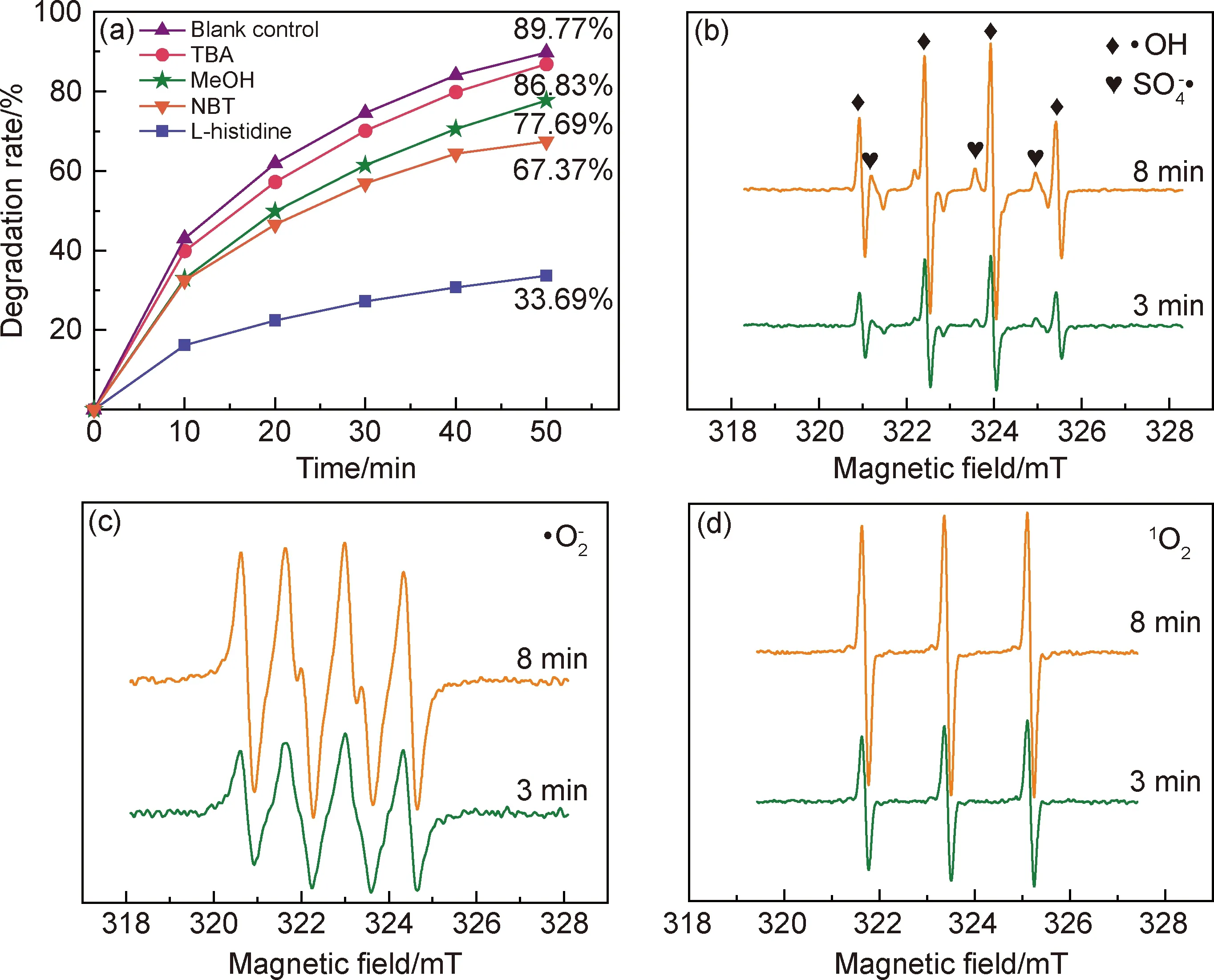
圖10 氧化活性中間體的鑒定結果
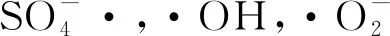
(8)
(9)
(10)
(11)
(12)
(13)
(14)
intermediates→H2O+CO2
(15)
3 結論
(1)在相同實驗條件下,當PMS和催化劑Co3O4,CoFe2O4和Fe2O3共同存在時,MB的降解率分別為42.08%,79.31%和20.53%。CoFe2O4表現出優于Co3O4和Fe2O3催化PMS降解MB溶液的能力,一方面源于其具有最高的比表面積和孔容,另一方面得益于兩種金屬離子之間的協同催化效應。
猜你喜歡
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
