HPLC-Q/Orbitrap HRMS快速篩查和確證液體乳中59種農藥殘留
2022-06-30 08:44:12張申平周靜杜茹蕓徐紅斌劉洋
中國乳品工業 2022年6期
張申平,周靜,杜茹蕓,徐紅斌,劉洋
(上海市質量監督檢驗技術研究院 國家市場監管重點實驗室(乳及乳制品檢測與監控技術),上 海200233)
0 引言
液體乳含有豐富的營養物質,種類繁多的液體乳產品備受消費者喜愛。但近年來液體乳中農藥[1-3]、獸藥[4-6]、有機污染物[7]殘留已經敲響了液體乳質量安全警鐘。液體乳的農藥殘留往往容易被忽視,這是因為飼料中的農藥經過奶牛消化吸收后,進入奶中的濃度較低,但這種微量、長期性的毒害勢必影響人體健康[8-9]。各國已然制定了液體乳中農藥殘留的最大限量值,歐盟規定了奶中460余種農藥的限量值,其中林丹的限量值低至0.001 mg/kg;美國規定了牛奶中190余種農藥的最大限量值,樂果的限量值低至0.002 mg/kg;日本規定了奶中300余種農藥的限量值,部分農藥一律不得檢出。我國的GB 2763-2021《食品安全國家標準 食品中農藥最大殘留限量》[10]已將牛乳中農藥殘留限量增加至125項。
液體乳中脂肪含量高、基質復雜、農藥含量低,因此需要更加高效的樣品前處理及儀器方法。目前主要的檢測方法有氣相色譜法[11-13]、氣相色譜-串聯質譜法(GC-MS/MS)[14-16]、液相色譜-串聯質譜法(LC-MS/MS)[17-19],主要集中在單一類別農藥的檢測。近年來高分辨質譜在農藥殘留分析方面的應用日益增多,高分辨質譜相對于常規HPLC-MS/MS具有更高的分辨率,在痕量組分的分析中具有獨特優勢,以一級質譜精確質量數、保留時間、同位素豐度比和二級特征離子為篩查條件,可以快速完成復雜樣品中痕量物質的篩查和確證。黃合田[20]等人建立了Sin-QuEChERS結合超高效液相色譜-四極桿-靜電場軌道阱高分辨質譜法快速非靶向篩查綠茶中38種農藥及其代謝物殘留的分析方法。孟志娟[21]等建立了氣相色譜-靜電場軌道阱高分辨質譜同時篩查農產品中70種農藥殘留的方法。吳潔珊[22]等人建立氣相色譜高分辨飛行時間質譜法快速篩查水果中283種農藥殘留量的分析方法。液體乳基質復雜,常規樣品提取液凈化方法難以實現痕量物質的分析,新型的Captiva EMR-Lipid小柱可通過體積排阻和疏水作用捕獲脂質,使體積較大的物質被阻止在外,達到凈化目的。張崇威[23]等人利用Captiva EMR-Lipid固相萃取結合超高效液相色譜-串聯質譜同時分析豬肉、雞肉、雞蛋中51種藥物殘留。胡巧茹[24]等人用Captiva EMR-Lipid小柱凈化,建立了超高效液相色譜-四極桿/靜電場軌道阱高分辨質譜快速篩查和確證糧谷產品中20種真菌毒素的方法。肖曉峰[25]等人對比分析了QuEChERS dSPE EMRLipid試劑盒和Captiva EMR-Lipid試劑盒對橄欖油食品模擬物中7種對苯二甲酸酯或苯甲酸酯的凈化效果。目前還未見Captiva EMR-Lipid小柱在液體乳農殘檢測中的應用報道。
本文利用Captiva EMR-Lipid小柱的超強凈化作用和靜電場軌道阱高分辨質譜建立液體乳中59種農藥殘留的液相色譜-高分辨質譜分析方法。涵蓋了有機磷類、有機氯類、擬除蟲菊酯類和氨基甲酸酯類等農藥;采用全掃描和二級質譜掃描相結合的方式,方便數據回溯;利用基質匹配標準溶液定量,降低了背景干擾,分析方法的各項參數可以滿足液體乳中農藥殘留的檢測要求。
1 材料與方法
1.1 儀器與試劑
Q Exactive PLUS四極桿-靜電場軌道阱高分辨質譜及Vanquish VF-P10-A高效液相色譜聯用系統,美國Thermo公司;電子天平,梅特勒-托利;5804離心機;渦旋振蕩器,德國heidolph;超純水器,MILLIPORE;Captiva EMR-Lipid凈化柱(6 mL,600 mg),美國Agilent公司;Thermo Accucore VDX色譜柱(100 mm×2.1 mm,2.6μm),美國Thermo公司;Cleanert S C18凈化柱和Cleanert MAS-Q dSPE凈化管,天津博納艾杰爾科技有限公司。
乙腈、甲醇(色譜純),美國Thermo公司;乙酸銨(分析純,≥98%)、甲酸(Aladdin,色譜級,≥98%),國藥集團化學試劑有限公司;萃取鹽包(QuEChERS Extract Pouch,EN Method,4 g硫酸鎂,1 g氯化鈉,1 g檸檬酸鈉二水合物,0.5 g檸檬酸氫鈉),美國Agilent公司。
農藥標準品:59種農藥混合標準品購自月旭上海科技有限公司,濃度50μg/mL(乙酸乙酯)。
1.2 樣品前處理
稱取5 g牛奶(精確至0.01 g)于50 mL離心管中,加入10 mL 0.2%甲酸-乙腈,同時加入萃取鹽包,渦旋混勻1 min,劇烈振蕩5 min。待離心管冷至室溫后,5 000 r/min離心2 min,取5 mL上清液于10 mL離心管中,加入1 mL超純水,輕微振蕩混勻。將混合液直接上樣至Captiva EMR-Lipid凈化柱,僅靠重力作用凈化,用1.5 mL乙腈-水(8∶2)混合溶劑沖洗凈化柱,結束后用洗耳球排空凈化柱。取3 mL流出液于40℃氮吹濃縮至1 mL,過0.22μm濾膜,上機測試。
1.3 標準溶液配制
基質匹配標準工作溶液配制:按照1.2的樣品前處理方法處理空白基質樣品,得到空白基質提取液,配制濃度為0.005、0.01、0.02、0.05、0.1 mg/kg的基質匹配標準工作溶液,該工作液應現配現用。
1.4 儀器條件
1.4.1 色譜條件
Thermo Accucore VDX色譜柱(100 mm×2.1 mm,2.6μm),柱溫:30℃,流速:0.4 mL/min,流動相A:5 mmol乙酸銨0.1%甲酸水溶液,流動相B:5 mmol乙酸銨0.1%甲酸甲醇溶液,梯度洗脫程序:0~4 min,0~20%B;4~5.5 min,20%~40%B;5.5~10.5 min,40%~100%B;10.5~12.9 min,100%B;12.9~15 min,100%~0 B。
1.4.2 質譜條件
離子源:可加熱電噴霧離子源(HESI);離子源溫度:325℃;噴霧電壓:3.60 kV;透鏡電壓:50.0 V;鞘氣流速:40 arb;輔助氣流速:10 arb;輔助氣溫度:350℃;質量分析器:Orbitrap;掃描模式:Full MS-ddMS2;采集范圍:120.0~1500.0 m/z;一級質譜分辨率為70 000 FWHM;C-trap最大容量(AGC target):1×106;C-trap最大注入時間:200 ms;二級質譜分辨率17 500 FWHM;C-trap最大容量(AGC target):2×105;C-trap最大注入時間:60 ms;歸一化碰撞能(NCE):20/40/60 eV;動態排除:8.0 s。
2 結果與討論
2.1 質譜條件的選擇
利用一級質譜全掃描的母離子精確質量數對應的色譜峰峰面積進行定量分析,全掃描范圍為120.0~1 500.0 m/z,利用保留時間和二級質譜的特征離子進行定性分析,根據響應強度,每個化合物指定兩個定性離子。黃合田[20]等人在高分辨質譜快速篩查綠茶中農藥及代謝物的研究中發現,乙硫甲威和滅蟲威的精確質量數相同,但可通過不同的保留時間和特征碎片離子(乙硫甲威:107.04927,滅蟲威:121.06472)進行區分。以Orbitrap作為質量分析器的質譜系統,其一級質譜的分辨率可達140 000,可以保證實測質量數和理論質量數之間的相對偏差足夠小,排除假陽性結果。孟志娟[21]等人報道當質譜分辨率低于60 000時,嘧霉胺的定性離子m/z 199.11049與干擾離子m/z 199.09990無法分開,容易造成假陽性結果。本研究中將一級質譜分辨率設置為70 000,二級質譜分辨率為17 500,相對質荷比偏差閾值設定為5×106。
2.2 色譜條件的優化
流動相的pH、是否添加緩沖鹽及不同的色譜柱對液相色譜分離化合物的效果至關重要。本研究比較了Thermo Accucore VDX色譜柱(100 mm×2.1 mm,2.6μm)和Thermo Accucore C18色譜柱(150 mm×2.1 mm,2.6μm)。結果表明,農藥化合物在兩根色譜柱上均有保留,但C18柱對部分化合物的響應略低,如四螨嗪和雙苯氟脲對應的峰面積僅為VDX柱峰面積的50%。流動相的pH及緩沖鹽影響目標化合物的離子化效率和分析靈敏度,分別考察了以甲醇、乙腈、5 mmol乙酸銨0.1%甲酸甲醇對目標物的分析靈敏度。發現以甲醇為流動相,基質匹配工作液中的氟蟲酰胺和丙硫菌唑未出峰;以乙腈作為流動相,咪唑菌酮和敵敵畏的響應值明顯降低,相比于5 mmol乙酸銨0.1%甲酸甲醇流動相,降低10倍。本研究采用梯度洗脫程序,15 min內,各化合物均能得到較好分離。基質匹配標準工作液中部分化合物的提取離子流圖如圖1所示。
2.3 前處理方法優化
在提取液中加入適量有機酸,有利于提高氨基甲酸酯類農藥的回收率[26-28]。本研究涉及的農藥極性范圍廣,提取液的選取對回收率至關重要。分別考察了乙腈、0.1%甲酸-乙腈、0.2%甲酸-乙腈對59種農藥的回收率,結果如圖2所示,隨著提取液中甲酸含量的增加,回收率<70%的農藥個數減少,回收率>120%的農藥個數增加,當提取液中甲酸濃度為0.2%時,回收率在70%~120%之間的農藥個數為51個,回收率<70%和回收率>120%的農藥個數分別為1和7個。
液體乳樣品提取液的凈化對農藥殘留檢測至關重要。本研究比較了Captiva EMR-Lipid凈化柱、Cleanert S C18凈化柱和Cleanert MAS-Q dSPE凈化管對液體乳樣品的凈化效果,采用Full MS模式采集凈化液的質譜信息。結果如圖3所示,Captiva EMRLipid凈化柱有效地隔絕了提取液中干擾物質的流出,凈化效果最優,而Cleanert MAS-Q dSPE凈化管幾乎沒有凈化效果。
2.4 基質效應
色譜分析中與目標物共流出的干擾組分會產生基質效應(Matrix Effect,ME),影響分析方法的靈敏度和定量結果的準確性。基質效應通常以目標物在基質匹配標準溶液和純溶劑標準溶液中響應值的差異程度進行評估,一種方法[29]是比較目標物峰面積,另一種方法[30]是比較基質匹配標準曲線的斜率和純溶劑標準曲線的斜率,后者可以更為全面地體現基質效應。本研究以基質匹配標準曲線斜率對純溶劑標準曲線斜率的相對偏差表示基質效應,作為ME值。若ME<10%,基質效應可忽略不計;ME值在10%~20%之間,則存在弱基質效應;ME值>20%,存在強基質效應。
實驗結果表明,54種農藥的ME≤10%,咪唑煙酸、滅蠅胺的ME>20%,多菌靈、氯氰菊酯、氟苯蟲酰胺的ME值在10%~20%之間,可見90%以上的農藥受基質影響較小。為進一步降低基質的影響,本研究采用基質匹配標準曲線定量。
2.5 方法評價
基質匹配標準曲線中以峰面積(y)對質量濃度(x)作圖,得到各農藥的標準曲線。各農藥化合物在0.005~0.1 mg/kg范圍內線性良好,相關系數R>0.99。對濃度為0.005 mg/kg的標準溶液進行逐級稀釋后進樣測試,根據3倍信噪比(S/N=3)估算檢出限(LOD),10倍信噪比(S/N=10)估算定量限(LOQ)。其中甲拌磷、塞普洛、甲氨基阿維菌素苯甲酸鹽B1A的定量限分別為0.012 mg/kg、0.0007 mg/kg和0.003 mg/kg,略高于GB 2763-2019國家標準中對牛乳規定的0.01 mg/kg、0.0004 mg/kg和0.002 mg/kg,其余農藥化合物的定量限均在0.0007~0.017 mg/kg之間,滿足國家標準中0.004~1 mg/kg的限量規定。加標回收實驗中3個加標濃度分別為0.005、0.02和0.05 mg/kg,每個濃度重復測定6次。以加標濃度為0.02 mg/kg的樣品為例,除雙甲脒回收率偏低,其余農藥化合物的回收率均在70%~130%之間,相對標準偏差(RSD)為2.54%~13.18%。
2.6 實際樣品分析
采用本研究建立的分析方法對20個市售液體乳樣品進行快速篩查與定量,樣品主要包含巴氏殺菌乳、超高溫滅菌乳、調制乳。篩查條件約束為質荷比偏差小于5×106、同位素豐度比大于90%、至少一個碎片離子與二級質譜數據庫匹配。兩個樣品中分別有丙溴磷、螺螨酯同時符合上述3個篩查條件,視為陽性檢出。利用基質匹配標準曲線進行定量分析,3次平行實驗中丙溴磷的檢出濃度為3.705、4.144、3.592μg/kg,螺螨酯檢出濃度為0.38、0.382、0.416μg/kg。我國國家標準GB 2763-2021規定牛乳中丙溴磷、螺螨酯最大限量值分別為10、4μg/kg,歐盟規定的最大限量值分別為10、4μg/kg,日本規定的最大限量值分別為10、10μg/kg,對比發現均未超標。陽性樣品中丙溴磷的色譜和質譜圖如圖4~6所示。
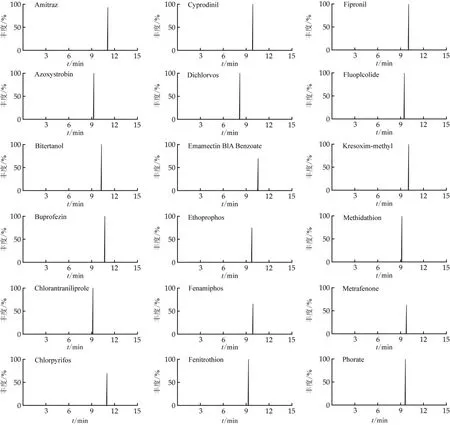
圖1 基制匹配標準工作液中部分農藥的提取離子流圖
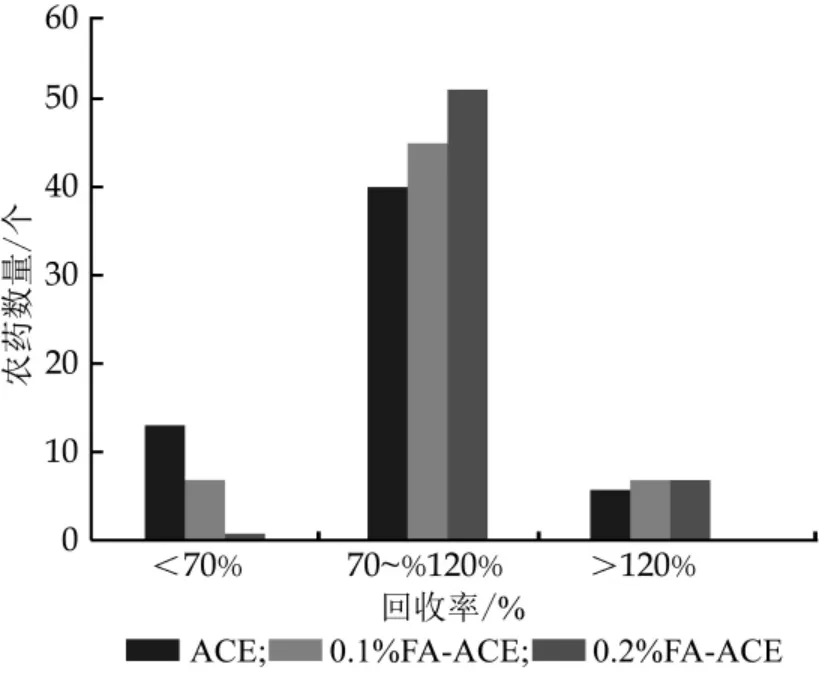
圖2 提取溶劑中甲酸含量對目標物回收率的影響
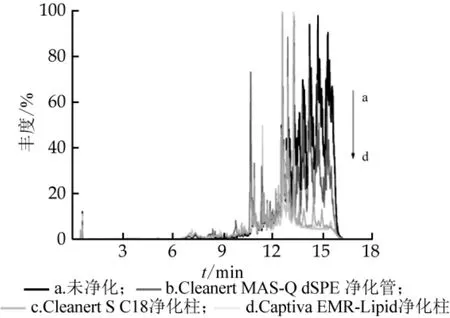
圖3 不同凈化方式下空白樣品的色譜圖
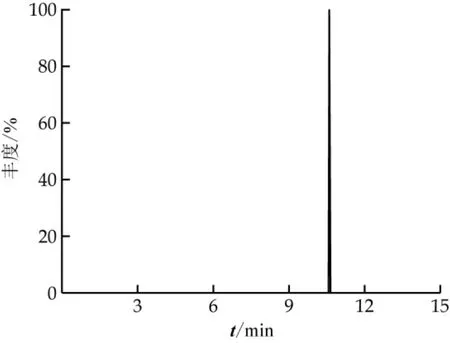
圖4 陽性樣品中丙溴磷色譜圖
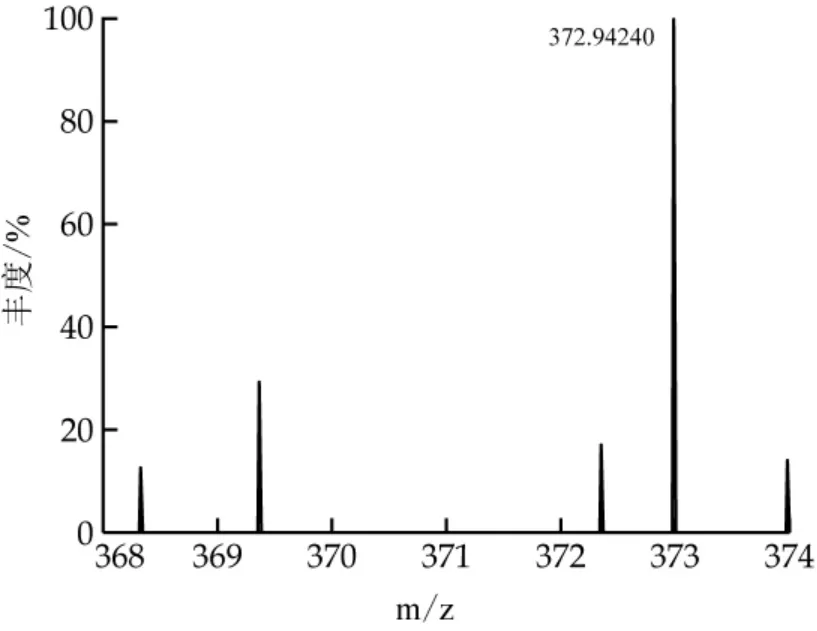
圖5 陽性樣品中丙溴磷一級質譜圖

圖6 陽性樣品中丙溴磷二級質譜圖
3 結論
本研究建立了液體乳中59種農藥殘留的高效液相色譜-四極桿-靜電場軌道阱高分辨質譜快速篩查與定量分析方法。樣品提取液經Captiva EMR-Lipid凈化柱一步凈化,簡化了凈化過程,提高了凈化效果;在15 min內可實現59種農藥殘留的色譜分離與質譜定量分析;利用自建農殘高分辨數據庫,以精確離子質量數、同位素豐度比、二級質譜特征碎片離子為篩查條件完成了快速篩查。在后續研究中還需要進一步擴展農藥高分辨質譜信息庫,將更多的農藥納入篩查范圍,尤其是極性較強、需使用特殊前處理方法和色譜條件的農藥分子,如矮壯素、敵草快、百草枯等。
