抑制線粒體鈣單向轉運體對大鼠急性胰腺炎氧化應激的作用研究*
2022-07-06 07:03:50覃穎穎楊慧瑩吳青謝金蓮蒙諾雷宇唐國都
中國現代醫學雜志 2022年12期
覃穎穎,楊慧瑩,吳青,謝金蓮,蒙諾,雷宇,唐國都
(1.廣西醫科大學第一附屬醫院 消化內科,廣西 南寧 530021;2.廣西醫科大學第二附屬醫院 消化內科,廣西 南寧 530007;3.右江民族醫學院第一附屬醫院 心血管內科,廣西 百色 533000)
急性胰腺炎(acute pancreatitis, AP)是臨床急腹癥的常見病因,20%~30%可進展為重癥[1],且進展兇險, 預后差[2]。 線粒體鈣單向轉運體(mitochondrial calcium uniporter, MCU) 是新近鑒定的位于線粒體內膜的特異性鈣離子通道,具有對釕紅(ruthenium red, RR)敏感、可順電化學梯度攝入Ca2+等特點[3]。胞內持續的鈣離子濃度升高可能會導致嚴重的細胞事件[4],引起細胞異常的代謝活動,最終導致細胞死亡[5-6]。氧自由基的參與是AP發病機制的重要環節,線粒體鈣信號可作為機體氧化信號的調節器[7]。強化抗氧化效應時可以改善AP 的嚴重程度[8],沉默信息調節因子3(silent information regulator 3, SIRT3)可通過多種途徑抑制機體生成活性氧(reactive oxygen species, ROS)[9]。DONG 等[10]研究發現,線粒體腔內的ROS 可以作為MCU 半胱氨酸97 敏感的信號源,促進MCU 高階低聚物形成,持續激活MCU 通道。本研究旨在探討抑制MCU、減輕線粒體鈣超載對AP 胰腺病理損傷和氧化應激的影響。
1 材料與方法
1.1 實驗動物
雄性Sprague-Dawley(SD)大鼠8~12 周齡、體重210~250 g,購于廣西醫科大學實驗動物中心,實驗動物生產許可證號:SCXK(桂)2020-003,實驗動物使用許可證號:SYXK(桂)2020-0004。大鼠在20~25℃空調房內飼養,通風良好,鼠籠定期清潔,自由進食、飲水。根據不同研究方法分為對照組、AP 組、AP+RR 組、RR 組,每組8 只。本研究經醫院醫學倫理委員會批準同意(No:2019-KY- 國基-017)。
1.2 主要試劑
雨蛙肽(上海Amquar 公司),Ⅳ型膠原酶、RR(美國Sigma公司),白細胞介素6(Interleukin-6,IL-6)酶聯免疫吸附試驗(enzyme linked immunosorbent assay, ELISA)試劑盒(武漢華美生物科技有限公司),丙二醛(Malondialdehyde, MDA)、還原型谷胱甘肽(Glutathione, GSH)檢測試劑盒(南京建成生物工程研究所有限公司),DHE 熒光探針(北京普利萊公司),Fluo-4 AM Ca2+(美國Thermo Fisher 公司)、Rhod-2 AM Ca2+熒光探針(美國Invitrogen 公司),βactin、MCU、SIRT3 兔源單克隆抗體、羊抗兔二抗(美國CST 公司),MnSOD 抗體(英國Abcam 公司)。
1.3 主要儀器
紅外線掃膜儀(美國Licor 公司),顯微鏡(日本Olympus 株式會社),多功能酶標儀(美國Thermo Fisher 公司),7600-120 型自動生化分析儀(日本HITACHI 株式會社),高速冷凍離心機(德國Eppendorf 公司)。
1.4 方法
1.4.1AP誘導大鼠禁食12 h,稱體重。對照組大鼠注射2.5 mg/kg 生理鹽水,AP 組大鼠腹腔注射50 μg/kg 雨蛙肽,每次間隔1 h,注射7 次,復制AP 模型;AP+ RR 干預組大鼠腹腔注射2.5 mg/kg RR,20 min 后腹腔注射50 μ g/kg 雨蛙肽,RR 組大鼠注射等量2.5 mg/kg RR。
1.4.2血清淀粉酶(Amylase, Amy)、脂肪酶(Lipase)活性和IL-6表達量測定最后1 次注射雨蛙肽結束開始計時。模型復制24 h 后于大鼠腹主動脈穿刺取血,4℃靜置30 min,3 500 r/min 離心10 min,取適量上清液,送至廣西醫科大學第一附屬醫院檢驗科檢測血清Amy、Lipase 活性;采用ELISA 試劑盒檢測血清IL-6 表達量,在多功能酶標儀450 nm 處讀取吸光度值。
1.4.3胰腺組織病理學評分模型復制24 h 后留取大鼠胰腺組織,在4%多聚甲醛中固定24 h,脫水,石蠟包埋。選取包埋組織4 μm 厚切片,蘇木精-伊紅(hematoxylin-eosin, HE)染色。顯微鏡下觀察每張樣本切片的5 個視野(×200)并評分,并取平均值,最終得分是每個病理參數的總和。胰腺組織病理學評分標準[11]見表1。
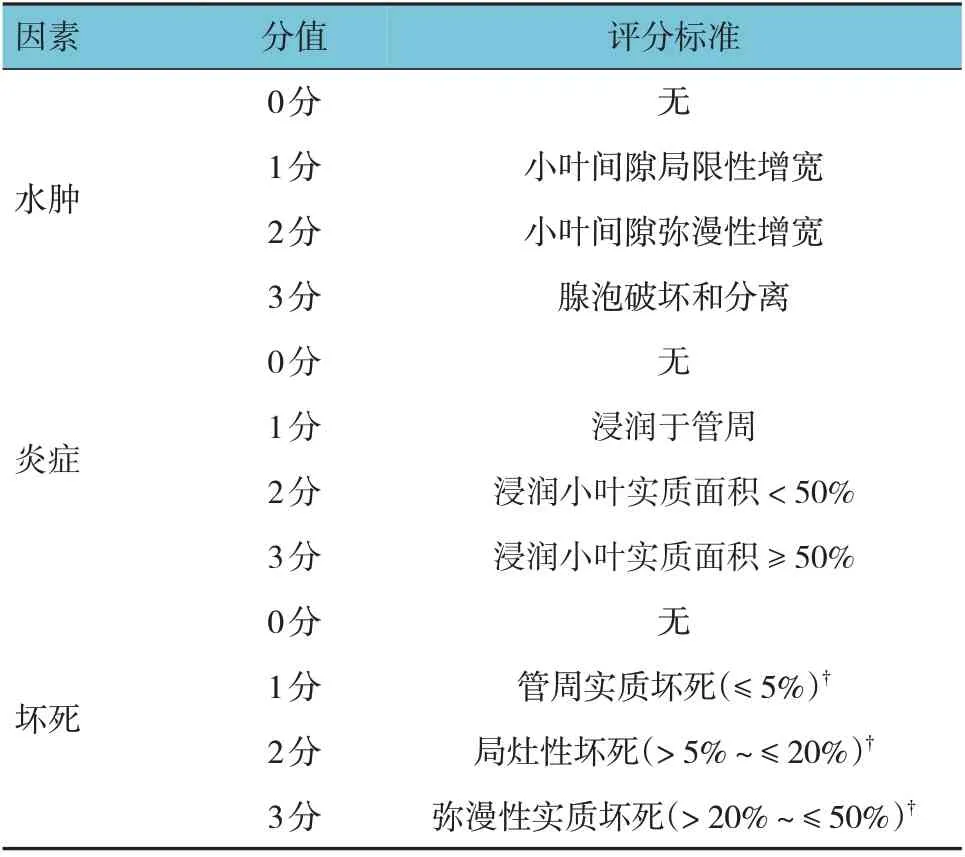
表1 胰腺組織病理學評分標準
1.4.4胰腺腺泡細胞線粒體超微結構觀察取2 mm×2 mm×2 mm 主胰管組織,戊二醛中避光固定>2 h,固定、脫水、滲透、包埋、切片、染色,電鏡(×2 500、×8 000)下隨機拍攝。
1.4.5腺泡細胞的提取及單細胞懸液的制備仔細切取主胰管周圍胰腺組織,大小約2 mm×2 mm×2 mm,剪碎,37℃環境下與1 mg/mL Ⅳ型膠原酶孵育15~30 min 后終止消化,用70 μm、40 μm 細胞過濾器過濾,1 000 r/min 離心5 min,杜氏磷酸鹽緩沖液(D-PBS)洗2、3 次后重懸。
1.4.6腺泡細胞線粒體內Ca2+、細胞內游離Ca2+、ROS 濃度檢測1 mL 腺泡單細胞懸液分別加入5 μmol/L Rhod-2 AM 和5 μmol/L Fluo-4 AM Ca2+探針工作液,按2∶3 體積在37℃黑暗中孵育30 min;1 mL 腺泡單細胞懸液和DHE 熒光探針按1 000∶1體積混勻,37℃避光孵育20 min。用D-PBS洗2、3 次后重懸,吸取500~800 μL 至35 mm×10 mm 細胞培養皿內,倒置熒光顯微鏡下觀察線粒體內Ca2+、細胞內游離Ca2+、ROS 濃度,曝光時間分別為32 ms、195 ms、32 ms。每個實驗重復3 次。
1.4.7胰腺組織GSH、MDA 含量的測定托盤天平稱取適量胰腺組織,胰腺組織與冰生理鹽水按重量(g)與體積(mL)1∶9 混合,冰浴后充分研磨,超聲裂解5 s,制成組織勻漿,4 000 r/min離心5 min,取上清液,按GSH、MDA 試劑盒說明書測定。
1.4.8Western blotting 檢測胰腺組織MCU、MnSOD、SIRT3 蛋白表達將蛋白樣品與5×上樣緩沖液(4∶1)混勻,煮沸5~10 min 備用。一抗稀釋濃度為1∶1 000,熒光二抗稀釋濃度為1∶10 000。轉膜條件:150 mA,1 h。每個實驗重復3 次。
1.5 統計學方法
數據分析采用SPSS 17.0 和GraphPad Prism 6.0統計軟件。計量資料以均數±標準差(±s)表示,比較用方差分析,進一步兩兩比較用LSD-t檢驗。P<0.05 為差異有統計學意義。
2 結果
2.1 各組大鼠胰腺組織病理評分和血清Amy、Lipase、IL-6水平比較
各組大鼠胰腺HE 評分和血清Amy、Lipase、IL-6 水平比較,經方差分析,差異有統計學意義(P<0.05),AP 組較對照組高(P<0.05),對照組與RR 組比較,差異無統計學意義(P>0.05),AP+RR干預組與AP 組比較,差異無統計學意義(P>0.05)。血清Amy、Lipase、IL-6 水平和組織學變化表明AP 模型復制成功。見圖1 和表2。
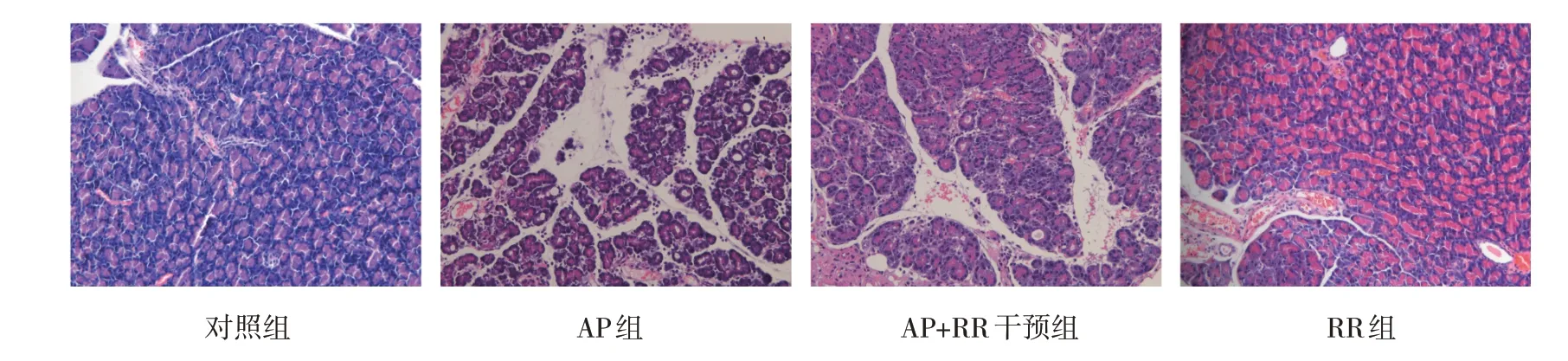
圖1 各組大鼠胰腺組織HE染色情況 (×200)
表2 各組大鼠胰腺組織病理評分和血清Amy、Lipase、IL-6比較 (n=8,±s)
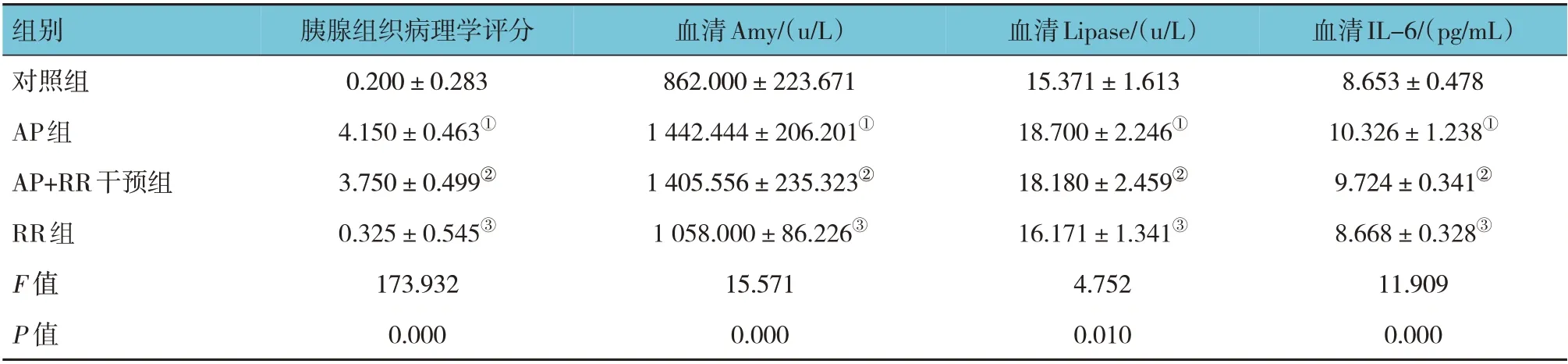
表2 各組大鼠胰腺組織病理評分和血清Amy、Lipase、IL-6比較 (n=8,±s)
注:①與對照組比較,P <0.05;②與AP組比較,P >0.05;③與對照組比較,P >0.05。
組別對照組AP組AP+RR干預組RR組F 值P 值血清IL-6/(pg/mL)8.653±0.478 10.326±1.238①9.724±0.341②8.668±0.328③11.909 0.000胰腺組織病理學評分0.200±0.283 4.150±0.463①3.750±0.499②0.325±0.545③173.932 0.000血清Amy/(u/L)862.000±223.671 1 442.444±206.201①1 405.556±235.323②1 058.000±86.226③15.571 0.000血清Lipase/(u/L)15.371±1.613 18.700±2.246①18.180±2.459②16.171±1.341③4.752 0.010
2.2 各組大鼠胰腺腺泡細胞超微結構
對照組、RR 組腺泡細胞整體結構尚可。AP 組腺泡細胞中度水腫,胞內局部電子密度減低,部分細胞器空泡變。細胞核呈不規則形,局部凹陷嚴重,異染色質邊集,提示細胞凋亡,核仁較大;線粒體中度腫脹、基質變淡、嵴斷裂、減少,少量嚴重者膜破損、基質外溢。AP+RR 組上述變化較AP 組減輕,腺泡細胞輕微水腫,細胞核偶見異染色質邊集,核膜完整;線粒體輕微腫脹,形狀、大小尚可,外膜模糊,嵴存在;余細胞器結構尚可。見圖2。
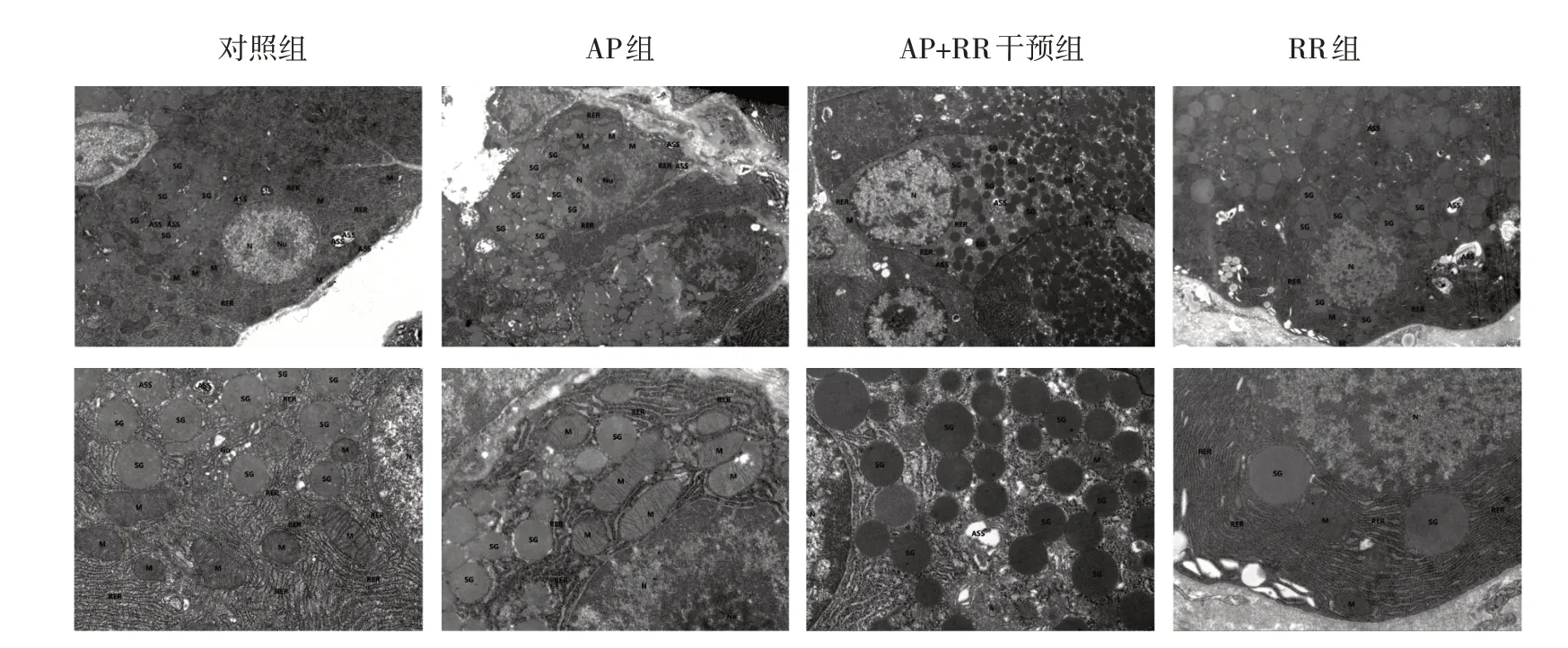
圖2 各組大鼠腺泡細胞超微結構的變化 (×8000)
2.3 各組大鼠MCU相對表達量,線粒體內Ca2+、細胞內游離Ca2+濃度比較
各組大鼠MCU 相對表達量,線粒體內Ca2+、細胞內游離Ca2+濃度比較,經方差分析,差異有統計學意義(P<0.05),AP 組高于對照組、RR 組(P<0.05),AP+RR 干預組低于AP 組(P<0.05)。見圖3~5 和表3。
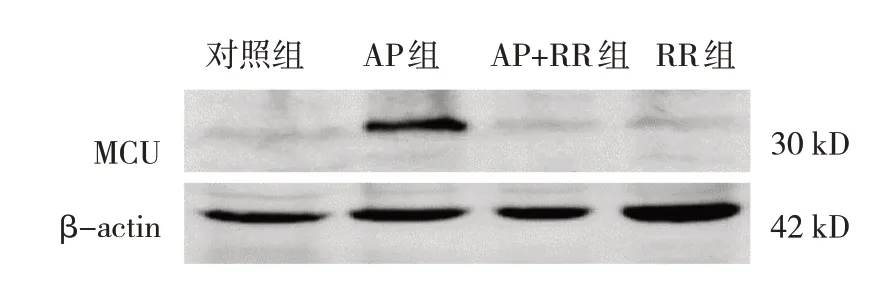
圖3 各組大鼠胰腺組織MCU蛋白的表達
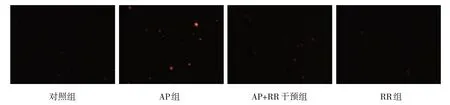
圖4 各組大鼠胰腺腺泡細胞線粒體內Ca2+熒光強度變化 (×200)
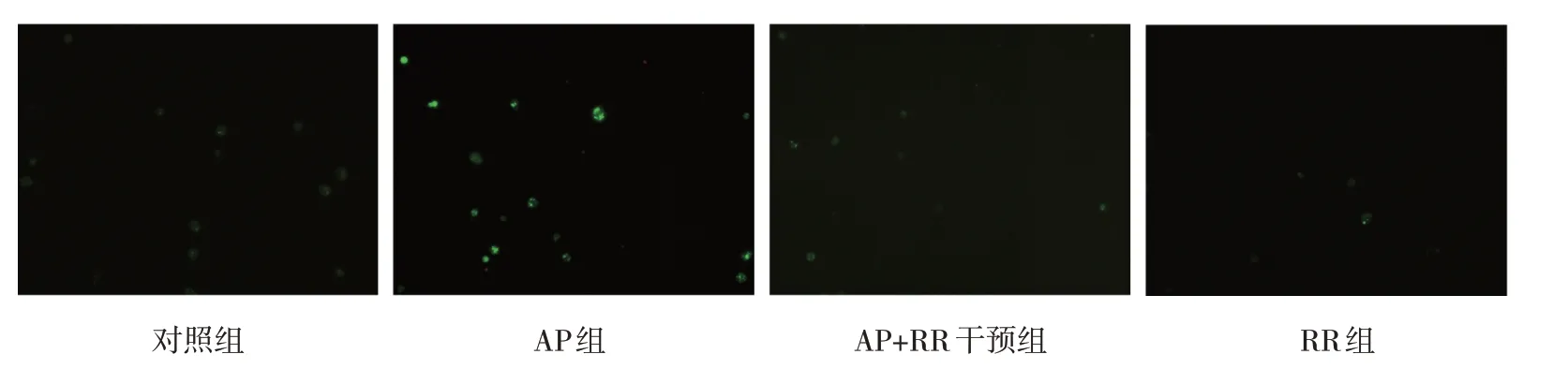
圖5 各組大鼠腺泡細胞內游離Ca2+熒光強度變化 (×200)
表3 各組大鼠MCU相對表達量,線粒體內Ca2+、胞內游離Ca2+濃度比較 (n=8,±s)
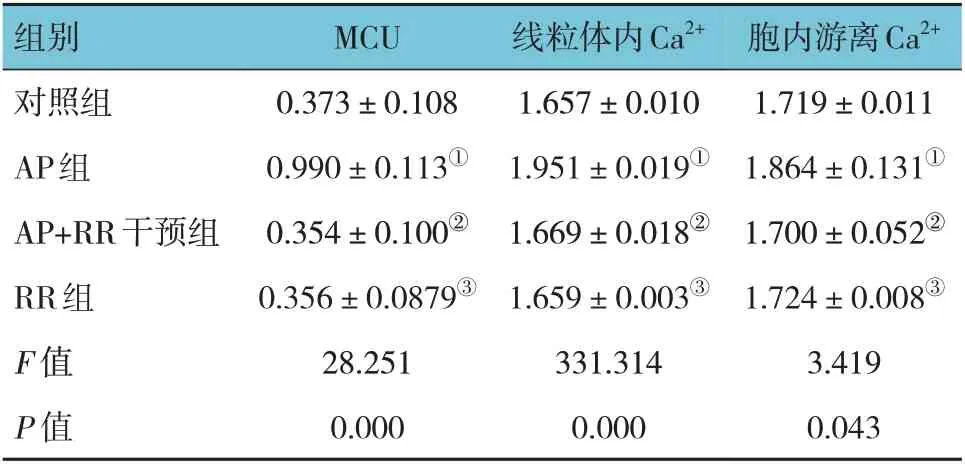
表3 各組大鼠MCU相對表達量,線粒體內Ca2+、胞內游離Ca2+濃度比較 (n=8,±s)
注:①與對照組比較,P <0.05;②與AP 組比較,P <0.05;③與對照組比較,P >0.05。
胞內游離Ca2+1.719±0.011 1.864±0.131①1.700±0.052②1.724±0.008③3.419 0.043組別對照組AP組AP+RR干預組RR組F 值P 值MCU 0.373±0.108 0.990±0.113①0.354±0.100②0.356±0.0879③28.251 0.000線粒體內Ca2+1.657±0.010 1.951±0.019①1.669±0.018②1.659±0.003③331.314 0.000
2.4 各組大鼠胰腺組織MnSOD、SIRT3 表達量,GSH、MDA含量和ROS熒光強度比較
各組大鼠胰腺組織MnSOD、SIRT3 表達量,GSH、MDA 含量和ROS 熒光強度比較,經方差分析,差異有統計學意義(P<0.05),AP 組MnSOD、SIRT3、GSH 低于對照組(P<0.05),ROS、MDA 高于對照組(P<0.05),AP+RR 干預組ROS、MDA 低于AP 組(P<0.05),MnSOD、SIRT3、GSH 高于AP 組(P<0.05)。見表4 和圖6~7。
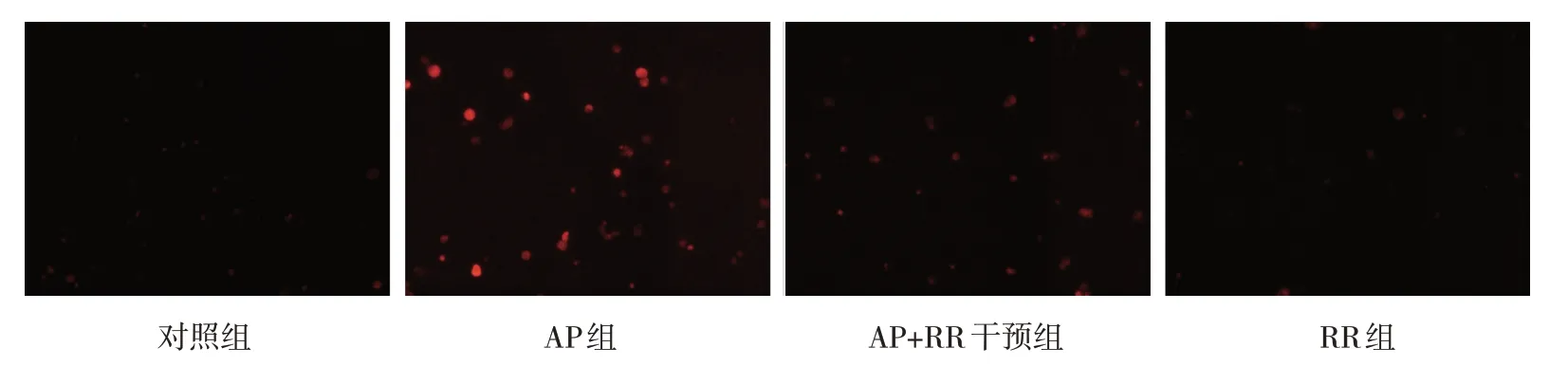
圖6 各組大鼠腺泡細胞ROS熒光強度變化 (×200)
表4 各組大鼠胰腺組織MnSOD、SIRT3表達量,GSH、MDA含量和ROS熒光強度比較 (n=8,±s)
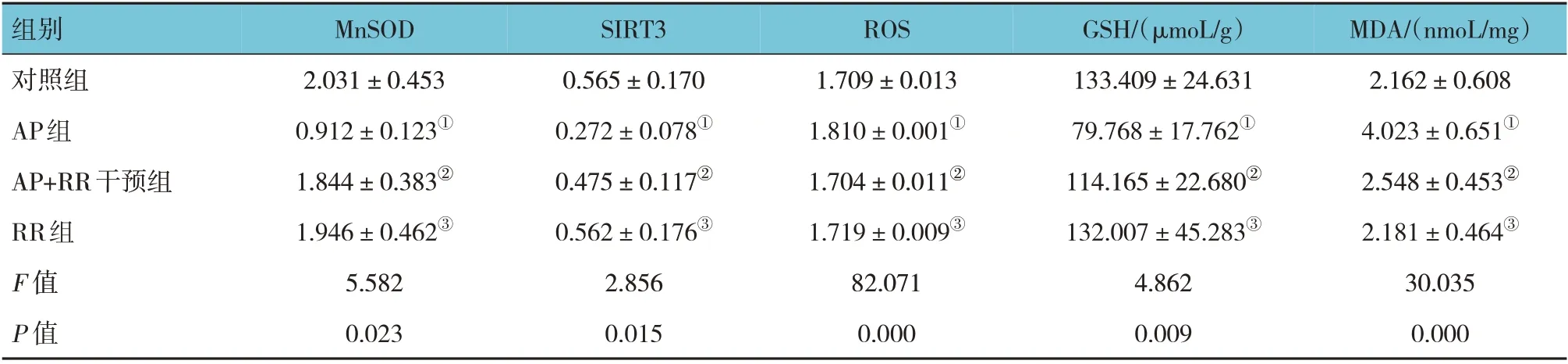
表4 各組大鼠胰腺組織MnSOD、SIRT3表達量,GSH、MDA含量和ROS熒光強度比較 (n=8,±s)
注:①與對照組比較,P <0.05;②與AP組比較,P <0.05;③與對照組比較,P >0.05。
組別對照組AP組AP+RR干預組RR組F 值P 值MDA/(nmoL/mg)2.162±0.608 4.023±0.651①2.548±0.453②2.181±0.464③30.035 0.000 MnSOD 2.031±0.453 0.912±0.123①1.844±0.383②1.946±0.462③5.582 0.023 SIRT3 0.565±0.170 0.272±0.078①0.475±0.117②0.562±0.176③2.856 0.015 ROS 1.709±0.013 1.810±0.001①1.704±0.011②1.719±0.009③82.071 0.000 GSH/(μmoL/g)133.409±24.631 79.768±17.762①114.165±22.680②132.007±45.283③4.862 0.009
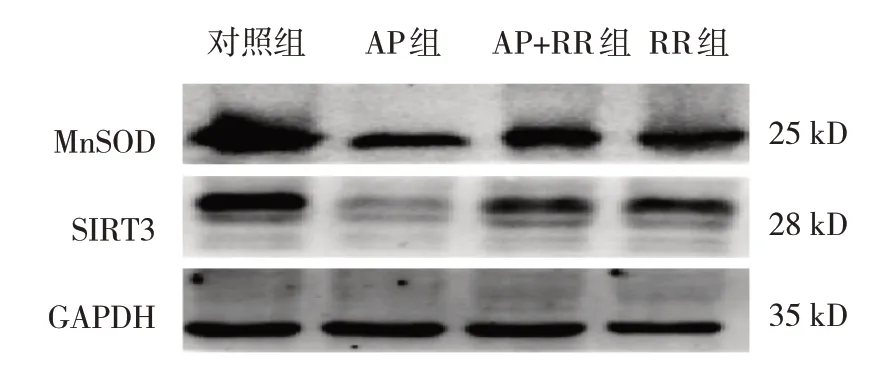
圖7 各組大鼠胰腺組織MnSOD、SIRT3蛋白的表達
3 討論
MCU 參與了多項生物學作用,其中在神經系統、心血管系統和腫瘤性疾病研究較多[12-15]。MCU通過Calpain/OPA-1 等通路,影響線粒體通透性轉換及穩態和線粒體分裂/融合的平衡,促進細胞凋亡,參與小鼠模型心肌缺血再灌注損傷和創傷性腦損傷[16-18];MCU 上調線粒體Ca2+攝取,通過抑制NAD+/SIRT3/SOD2 通路和ROS 激活的c-jun 氨基末端激酶途徑,促進基質金屬蛋白酶-2 的活性[19]。激活ROS/NF-κB 信號,抑制轉錄因子A-線粒體磷酸化促進線粒體的生物發生[20],促進腫瘤細胞的轉移和體內外生長。線粒體是經MCU 運輸的鈣離子的接收器,線粒體鈣離子信號異常會導致線粒體形態完整性發生變化。AP 起病過程中,腺泡細胞鈣離子內流和氧化應激反應增強,線粒體形態以及結構的異常。而應用MCU 的抑制劑RR 后,可觀察到腺泡細胞超微結構損害減輕,鈣內流減弱,抗氧化效應增強。這為AP 鈣超載、線粒體損傷、氧化應激提供了生化指標和形態學證據。
Western blotting 和熒光探針結果表明,在雨蛙肽誘導的AP 大鼠中,MCU 介導的氧化應激損傷導致氧化指標ROS 和MDA 水平上升,氧化保護因子MnSOD、GSH 水平下降。有趣的是,SIRT3 在MCU 介導的氧化應激損傷中也呈低表達,而當MCU 的作用被RR 抑制后,氧化應激水平下降,SIRT3 的蛋白表達水平上升,這提示SIRT3 可能是AP 氧化應激損傷的保護因素,MCU 的激活可能抑制了SIRT3 介導的相關通路,誘發氧化應激。MCU 的活化可以抑制肝癌細胞中的NAD+/SIRT3/SOD2 通路,促進肝癌細胞產生ROS,增強肝癌細胞的體外侵襲和遷移能力[17],表明SIRT3 的活化可能抑制MCU 介導的ROS 水平,從而抑制氧化應激的發生、發展,與本研究結果相符。SIRT3 可能是治療AP 氧化應激損傷的關鍵靶點。
不幸的是,抑制MCU 無法改善AP 的病理嚴重程度。這一結果與CHVANOV 等[21]結果是一致的。研究表明,線粒體鈣超載并不是AP 誘導劑引起的唯一損傷效應,其可能僅參與腺泡細胞損傷和AP 啟動的部分過程。即使是在MCU/CYD-D雙基因敲除小鼠中,MCU基因完全缺失,實驗動物的腦線粒體仍能攝取Ca2+[17]。DIA 等[22]發現,16 周的高脂肪高蔗糖(high-fat high-sucrose diet, HFHSD)喂養的小鼠出現胰島素抵抗、糖尿病心肌病,在HFHSD 心肌細胞中觀察到IP3R/Grp75/VDAC 鈣通道復合體減少,IP3 刺激鈣向線粒體的轉運減少,但是MCU 及其調節亞基MICU1 并未參與到線粒體鈣離子紊亂這一過程中,HFHSD 心肌細胞線粒體鈣攝取減少主要是由于網狀-線粒體功能性鈣耦聯減少所致。本研究發現抑制MCU 可能通過SIRT3/MnSOD 通路來抑制早期胰腺炎的氧化應激反應,但同樣也顯示了RR 并不能顯著改善胰腺炎的病理嚴重程度。MCU 在AP 的發生、發展過程中的作用可能屬于“微效”蛋白。然而在臨床中,即使是輕癥胰腺炎患者的恢復時間也需要1 周以上,AP 的臨床治療過程則更加漫長。有研究指出,在內皮細胞功能障礙中,高糖誘導的MCU相關的鈣超載、凋亡和ROS 損傷的表現呈劑量和時間依賴性[23],RR 在AP 的治療后期仍有巨大的潛在價值。
猜你喜歡
中老年保健(2021年3期)2021-08-22 06:50:04
昆明醫科大學學報(2021年1期)2021-02-07 01:06:36
現代臨床醫學(2021年1期)2021-01-26 00:56:02
中華養生保健(2020年4期)2020-11-16 01:31:40
中西醫結合肝病雜志(2020年2期)2020-10-27 02:18:50
世界科學技術-中醫藥現代化(2020年2期)2020-07-25 02:05:56
西南軍醫(2016年6期)2016-01-23 02:21:19
新疆醫科大學學報(2015年10期)2015-12-26 12:33:30
癌變·畸變·突變(2015年3期)2015-02-27 06:15:09
西南軍醫(2015年2期)2015-01-22 09:09:37
