可磁性回收鈀催化劑的制備及其催化Suzuki偶聯反應1
2022-07-06 05:37:30林小敏馬逸青柯家曉馬浩喬艷輝滕俊江李凝
廣東石油化工學院學報 2022年3期
關鍵詞:催化劑
林小敏,馬逸青,柯家曉,馬浩,喬艷輝,滕俊江,李凝
(1.廣東石油化工學院 化學學院,廣東 茂名 525000; 2.廣東石油化工學院 化學工程學院,廣東 茂名 525000)
鈀(Pd)催化的Suzuki偶聯反應是制備聯苯結構單元化合物的重要方法,已成為構建“碳-碳”(C—C)鍵的關鍵有機單元反應之一,在精細化工、藥物、農藥及有機功能材料等領域中得到廣泛應用[1]。
Pd是貴金屬,儲量稀少,如何提高Suzuki偶聯反應中Pd催化劑的回收使用,成為該領域重點研究的內容之一。將催化劑固體化,制備多相Pd催化劑,成為提高Pd回收利用的有效方式[2]。在Pd基多相催化劑中,磁性內核的引入,更簡化了Pd催化劑的回收使用方法[3]。近年來,以Fe3O4、γ-Fe2O3等為內核的可磁性回收Pd催化劑的制備受到科研工作者的廣泛關注,并取得較大進展[4]。為了穩定催化劑活性中心(Pd(0)),利用含有孤對電子的氮(N)、磷(P)、氧(O)等配原子的配體基團對磁性內核進行修飾是制備高效、穩定、可磁性回收Pd催化劑的重要手段。然而,該類方法仍存在操作過程繁瑣、使用毒性大的化學試劑等不足,嚴重影響磁性Pd催化劑在Suzuki偶聯反應中的應用[5]。因此,在碳中和、碳達峰的背景下,如何簡單、高效地構建可磁性回收的Pd催化劑,對于Suzuki偶聯反應的應用和發展具有重要意義。
淀粉是無毒、可再生的天然高分子材料,含有大量的羥基(—OH)官能團,對Pd催化劑活性中心同樣具有一定的穩定作用。基于此,本文以Fe3O4為磁性內核,以淀粉為Pd的穩定劑,制備淀粉包覆Fe3O4的磁性材料,隨后將Pd催化劑負載于淀粉穩定層,構建可磁性回收的Pd催化劑Fe3O4@Starch-Pd(0),并系統研究其催化Suzuki偶聯反應的性能和回收使用特性。
1 實驗部分
1.1 儀器與試劑
儀器:傅里葉變換紅外光譜儀(FT-IR,Nicolet 6700,美國賽默飛世爾科技公司);X射線粉末衍射儀(XRD,Ultima IV,日本理學);同步熱分析儀(TGA,STA449F3,德國耐馳公司);電感耦合等離子體發射光譜儀(ICP,iCAP7600,賽默飛世爾科技公司);核磁共振波譜儀(NMR,DRX400,德國布魯克公司)。
試劑:氯化鈀(PdCl2,分析純)、鹵代芳烴(分析純)、芳基硼酸(分析純)等購自上海達瑞精細化學品有限公司;可溶性淀粉(分析純)等其他試劑購自光華科技股份有限公司,且均未進一步純化處理。
1.2 催化劑的制備及表征
Fe3O4@Starch-Pd(0)催化劑制備步驟:(1)根據文獻[6]報道的方法制備Fe3O4納米粒子,保存備用;(2)向20 mL蒸餾水中加入1.0 g可溶性淀粉,待淀粉完全溶解后,加入0.1 g Fe3O4納米粒子,超聲30 min,形成穩定的磁性懸浮液;(3)將上述磁性懸浮液緩慢倒入250 mL的無水乙醇中,機械攪拌,淀粉析出,形成淀粉包覆的磁性絮狀物Fe3O4@Starch,磁性分離后,保存備用;(4)將0.15 g PdCl2溶解于30 mL無水乙醇中,隨后加入上述磁性絮狀物(Fe3O4@Starch),機械攪拌,在60 ℃的條件下反應12 h,即可獲得以Fe3O4為磁性內核,淀粉包覆層穩定的Fe3O4@Starch-Pd(0)催化劑,磁性分離后,洗滌、真空干燥,保存備用;(5)使用FT-IR、XRD對催化劑的結構進行表征,利用TGA對催化劑的熱穩定性進行評價,同時采用ICP對催化劑中Pd含量進行分析。
1.3 Fe3O4@Starch-Pd(0)催化Suzuki偶聯反應
向25 mL的圓底燒瓶中加入對硝基碘苯(1.0 mmol,0.249 g)、苯硼酸(1.1 mmol,0.134 g)、碳酸鉀(2.0 mmol,0.276 g)、無水乙醇(EtOH,5.0 mL)及催化劑(0.05 g),磁力攪拌,加熱回流,用薄層色譜法(TLC)追蹤反應進程。反應結束后,通過外加磁鐵的方式將催化劑與反應液分離,收集反應液后,回收的催化劑可直接重復使用。向上述收集的反應液中加入0.5 mL的蒸餾水,放置,在乙醇揮發過程中,產物以晶體的形式析出,待乙醇揮發完后(結晶過程),過濾、水洗、干燥后即可得到目標產物。最后,通過1H NMR和13C NMR對其結構進行鑒定(均為已知物)。
2 結果與討論
2.1 催化劑結構表征
通過FT-IR對催化劑的分子結構進行描述,結果見圖1。波數為3600~3100 cm-1的吸收峰對應于—OH的伸縮振動,3000~2800 cm-1的吸收峰對應于亞甲基(—CH2—)的C—H伸縮振動,1680 cm-1的吸收峰則歸屬于—OH(淀粉、H2O)的彎曲振動,1450 cm-1的吸收峰為亞甲基的C—H彎曲振動,1190 cm-1的吸收峰為C—O的伸縮振動,920 cm-1的吸收峰為C—O—C骨架伸縮振動,573 cm-1則為Fe—O的伸縮振動的吸收峰。對比圖1中曲線a、b、d可看出,Fe3O4@Starch-Pd(0)催化劑中含有淀粉和Fe3O4結構,表明成功制備了淀粉包覆Fe3O4內核的磁性材料。隨后,通過XRD對催化劑的晶體結構進行分析,結果見圖2。2θ位于30.14°、35.60°、43.33°、53.54°、57.15°、62.96°的衍射峰分別對應于Fe3O4的(220)、(311)、(400)、(422)、(511)、(440)晶面;2θ位于40.01°、46.57°、67.95°的衍射峰則分別歸屬于Pd(0)的(111)、(200)、(220)晶面。2θ介于12°~25°的寬峰為無定型結構淀粉的衍射峰。通過對比圖2曲線a、b、d可知,所制備的催化劑中同時含有Fe3O4、淀粉以及Pd(0)組分。結合FT-IR結果(圖1),證明成功制備了磁性Fe3O4@Starch-Pd(0)催化劑。此外,通過ICP分析發現Fe3O4@Starch-Pd(0)催化劑中Pd的負載量為0.38 mmol/g。
Suzuki偶聯反應常在加熱的條件下進行,因此Fe3O4@Starch-Pd(0)催化劑的熱穩定性也是考察的重要內容。從圖3中曲線a可知,Fe3O4的結構較穩定,熱穩定性好,隨溫度的升高,其熱損失較少。而催化劑Fe3O4@Starch-Pd(0)在100 ℃之前有10%左右的質量損失(曲線b),這主要是因為樣品吸收的少量水分所致。隨后該催化劑在230 ℃時出現明顯的質量損失,并于290~340 ℃內出現快速失重現象,分別歸因于樣品中無定型及結晶淀粉組分的分解反應,說明該催化劑的使用溫度上限為230 ℃。通過對比Fe3O4@Starch-Pd(0)與淀粉的熱分解曲線(a、d)發現淀粉的熱分解開始溫度為270 ℃,高于Fe3O4@Starch-Pd(0)中淀粉的開始分解溫度,且淀粉的快速熱分解溫度與Fe3O4@Starch-Pd(0)中的淀粉相當。這一現象主要是因可溶性淀粉具有一定的晶體結構(圖2,曲線d),具有較好的熱穩定性。在催化劑制備過程中,淀粉溶解、再生時會有一部分轉化為無定型結構(圖2,曲線b),在一定程度上降低了其熱穩定性。此外,當加熱溫度升至800 ℃,Fe3O4@Starch-Pd(0)的失重率為73%(曲線b),而可溶性淀粉的失重率為84%(曲線d),Fe3O4失重率約為7%,充分說明所制備的磁性Pd催化劑中含大量的淀粉組分,進一步證實成功制備了磁性Pd催化劑。Fe3O4@Starch-Pd(0)催化劑的熱分解曲線圖表明該催化劑具有較好的熱穩定性。
2.2 催化Suzuki偶聯反應
2.2.1 優化反應條件
本文以對硝基碘苯與苯硼酸之間的Suzuki偶聯反應為模型,研究Fe3O4@Starch-Pd(0)催化劑用量、溶劑組成、堿的類型以及反應溫度對該反應的影響規律,見表1。模型:
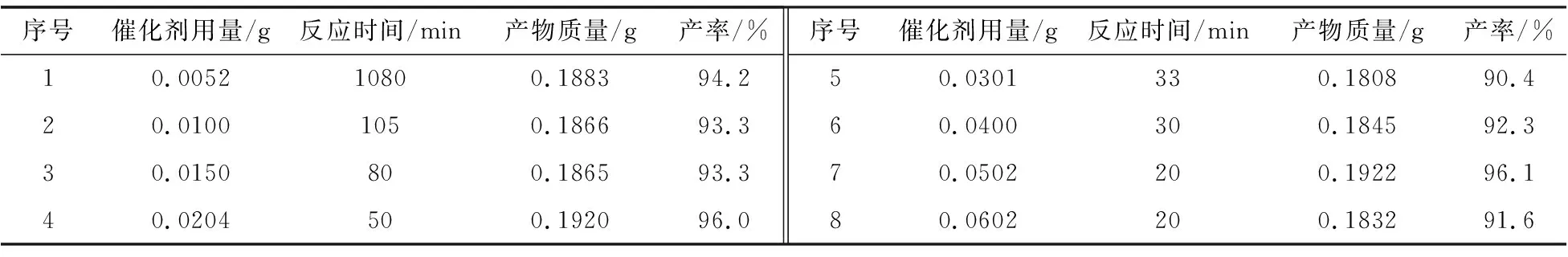
表1 催化劑的用量對Suzuki反應的影響
從表1中可知,催化劑用量對該反應有明顯的影響。隨著催化劑用量的增加,該模型反應目標產物的產率幾乎相同,均在90%以上,但完全反應的時間逐漸縮短,體現Pd(0)活性中心對該模型反應的催化作用[7]。當催化劑用量增加到0.05 g時,完全反應時間縮短到20 min;繼續增加催化劑用量,完全反應時間沒變,表明最佳的催化劑用量為0.05 g(1.9 mol% Pd)。
溶劑對反應體系中堿的溶解具有較大的影響,從而影響Suzuki反應的性能。由于淀粉包覆層溶于水,因此本文選擇非水相的溶劑體系,結果如表2所示。從表中可以看出,無水乙醇(EtOH)為最佳溶劑,其所對應目標產物的產率遠高于EtOH(95%)、DMF、丙酮以及四氫呋喃體系。因此,本文后續反應選擇EtOH為最佳溶劑。
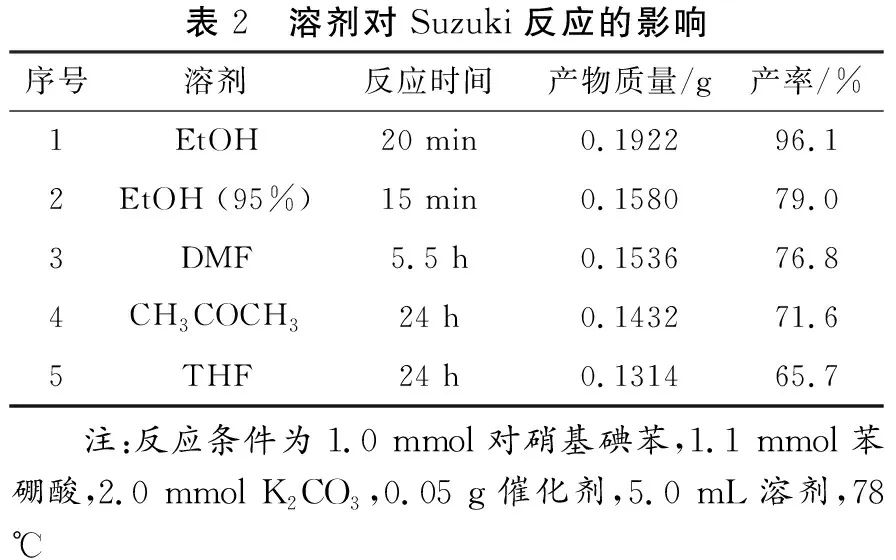
表2 溶劑對Suzuki反應的影響序號溶劑反應時間產物質量/g產率/%1EtOH20 min0.192296.12EtOH (95%)15 min0.158079.03DMF5.5 h0.153676.84CH3COCH324 h0.143271.65THF24 h0.131465.7 注:反應條件為1.0 mmol對硝基碘苯,1.1 mmol苯硼酸,2.0 mmol K2CO3,0.05 g催化劑,5.0 mL溶劑,78 ℃
從Pd催化的Suzuki偶聯反應機理可知,堿作為芳基硼酸的活化劑,對Suzuki偶聯反應的發生起到決定性作用。由表3可知,不同種類的堿對Suzuki偶聯反應均具有較好的促進作用。然而,強堿(NaOH、KOH)腐蝕性太強,而Na3PO4·12H2O穩定性較差,Cs2CO3成本較高,因此從經濟性、安全性及催化效率的角度考慮,選擇K2CO3為后續反應的堿。Pd催化的Suzuki偶聯反應同時對反應溫度較敏感,因此本文繼續考察反應溫度對該催化反應的影響規律。由表4可知,在Fe3O4@Starch-Pd(0)催化劑的作用下,Suzuki偶聯模型反應在室溫下即可進行,目標產物的產率達到95.0%,但完全反應時間較長(12 h)。當反應溫度升高時,產率幾乎不變,完全反應時間明顯縮短。當達到乙醇的回流溫度(沸點,78 ℃)時,該模型反應在20 min內即可進行完全。因此,選擇乙醇的回流溫度(沸點)為最佳的反應溫度。綜上所述,最佳的催化劑用量為0.05 g,溶劑為EtOH,堿為K2CO3,反應溫度為78 ℃。
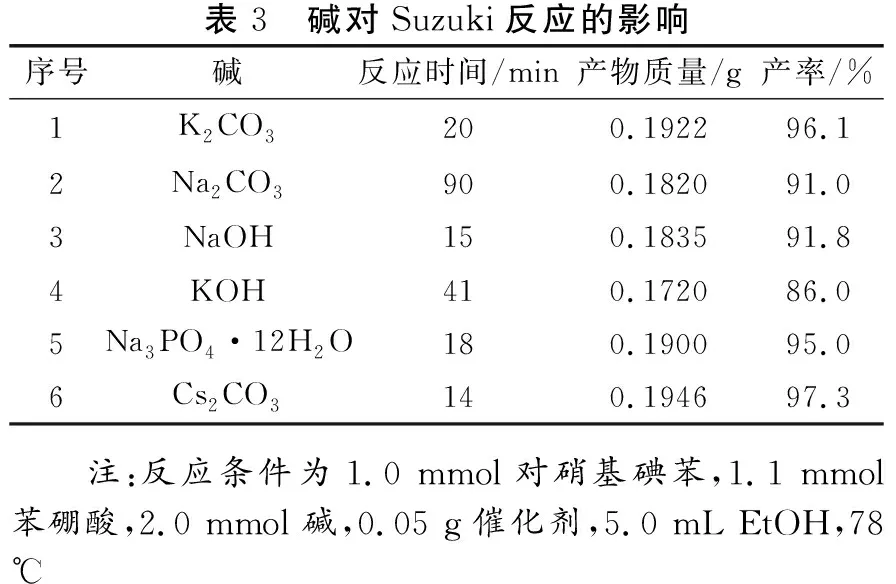
表3 堿對Suzuki反應的影響序號堿反應時間/min產物質量/g產率/%1K2CO3200.192296.12Na2CO3900.182091.03NaOH150.183591.84KOH410.172086.05Na3PO4·12H2O180.190095.06Cs2CO3140.194697.3 注:反應條件為1.0 mmol對硝基碘苯,1.1 mmol苯硼酸,2.0 mmol 堿,0.05 g催化劑,5.0 mL EtOH,78 ℃
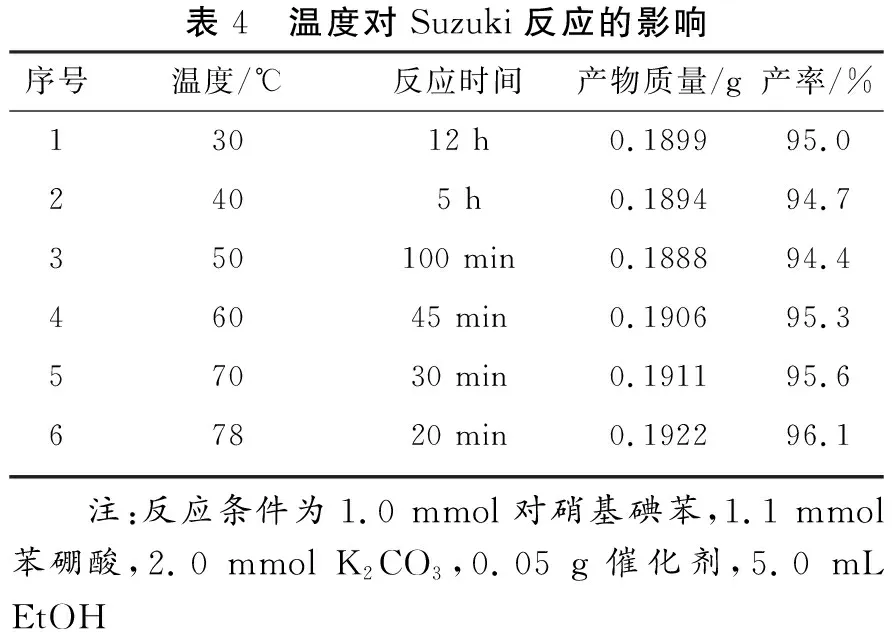
表4 溫度對Suzuki反應的影響序號溫度/℃反應時間產物質量/g產率/%13012 h0.189995.02405 h0.189494.7350100 min0.188894.446045 min0.190695.357030 min0.191195.667820 min0.192296.1 注:反應條件為1.0 mmol對硝基碘苯,1.1 mmol苯硼酸,2.0 mmol K2CO3,0.05 g催化劑,5.0 mL EtOH
2.2.2 底物拓展
為了進一步評價Fe3O4@Starch-Pd(0)催化劑的實用性,本文繼續對反應底物進行拓展,系統研究該催化劑對催化不同結構的鹵代芳烴和芳基硼酸之間的Suzuki偶聯反應的普適性,結果見表5和表6。
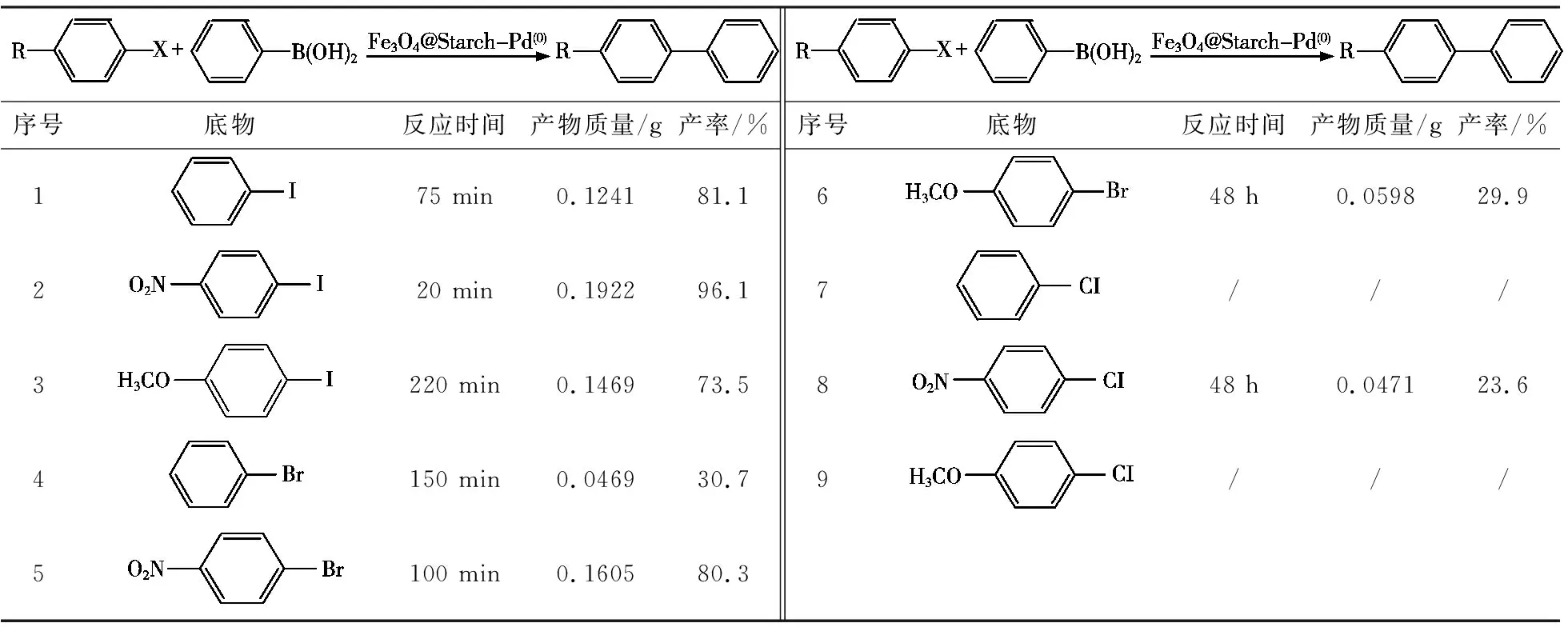
表5 鹵代芳烴底物拓展
從表5可知,在Fe3O4@Starch-Pd(0)催化劑的作用下,鹵代芳烴中鹵素種類對其Suzuki偶聯反應效率具有較大影響。催化碘苯和苯硼酸之間的偶聯反應效率遠高于溴苯(表5,序號1和4)。主要因為溴苯中C—Br的鍵能比碘苯中C—I的鍵能高,即碘取代基比溴取代基易于脫去[7]。C—Cl鍵能更強,因此以氯苯為底物時,則不能進行Suzuki偶聯反應形成聯苯產物(表5,序號7)。整體而言,鹵代芳烴反應發生難易順序為—I>—Br>—Cl。
此外,鹵代芳烴的取代基對其與苯硼酸之間的偶聯反應同樣具有較大影響。含有吸電子基團的鹵代芳烴更易進行Suzuki偶聯反應,且產率較高(表5,序號2和5);而含有供電子基團的鹵代芳烴反應活性明顯降低(表5,序號3和6)。主要是由于吸電子基團可以有效降低碳-鹵鍵(C—X)的鍵能,從而提高相應活性[7,8];而供電子基團則增強了C—X的鍵能,不利于其進行Suzuki偶聯反應[5]。借助于吸電子基團的活化作用,反應活性極低的氯苯經過硝基功能化后(對硝基氯苯),可在Fe3O4@Starch-Pd(0)催化劑的作用下與苯硼酸進行偶聯反應,產率達到23.6%(表5,序號8)。與鹵代芳烴對該反應的影響相比,不同取代的芳基硼酸對該反應催化轉化效率影響不大,產率可達73.9%~96.1%。因此,通過對芳基硼酸進行功能化,有望成為合成含功能取代基聯苯化合物的重要手段。
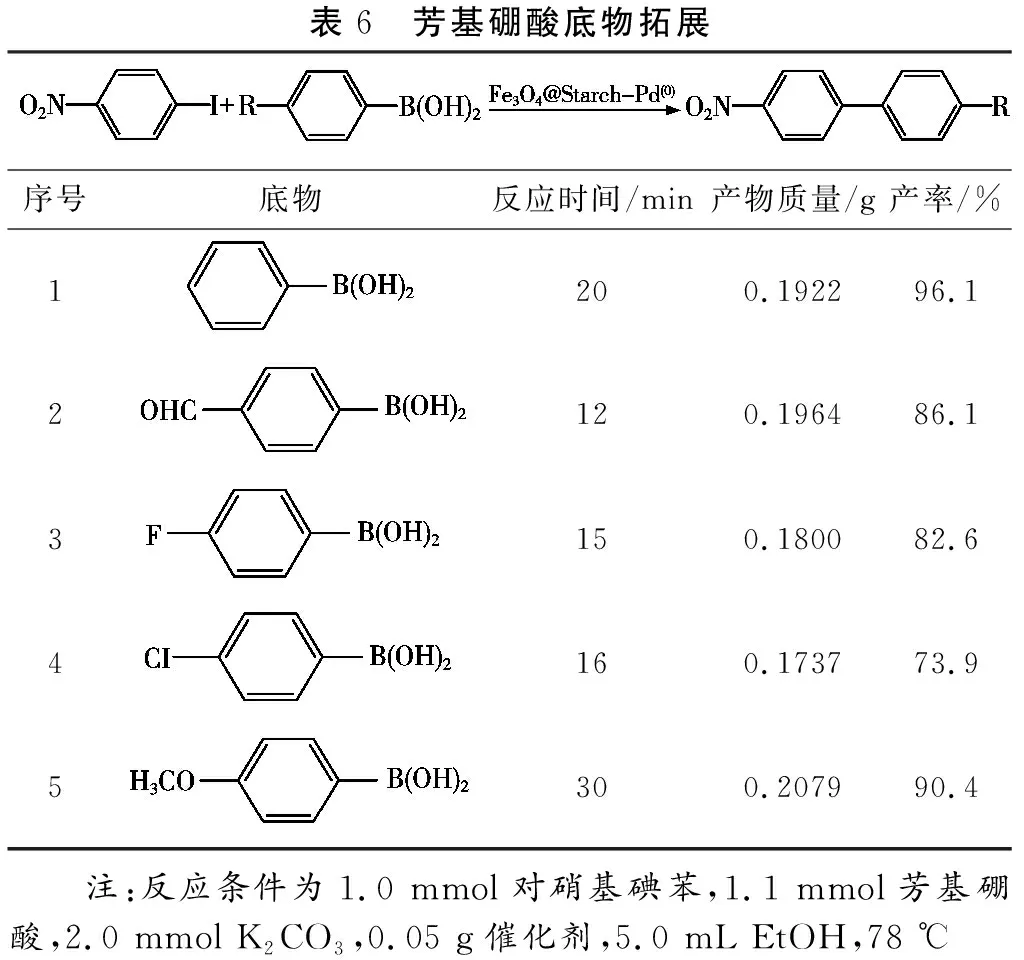
表6 芳基硼酸底物拓展序號底物反應時間/min產物質量/g產率/%1200.192296.12120.196486.13150.180082.64160.173773.95300.207990.4 注:反應條件為1.0 mmol對硝基碘苯,1.1 mmol芳基硼酸,2.0 mmol K2CO3,0.05 g催化劑,5.0 mL EtOH,78 ℃
綜上所述,對鹵代芳烴和芳基硼酸的底物進行了拓展,生成了系列功能化聯苯產物,證明Fe3O4@Starch-Pd(0)催化劑具有一定的底物普適性。
2.3 催化劑回收使用
簡單高效的磁性回收方式是該催化劑的重要特色,本文通過磁性分離的方式對Fe3O4@Starch-Pd(0)
催化劑進行回收使用,實驗結果見表7。從表中可知,催化劑在循環使用過程中,隨著重復使用次數的增加,目標產物仍然保持著較好的產率,但Suzuki偶聯反應時間逐漸延長,說明催化劑活性呈現逐漸下降的趨勢。尤其是循環使用4次后,完全反應時間增加到100 min以上(表7,次數5和6),說明催化劑重復使用4次后活性明顯下降。
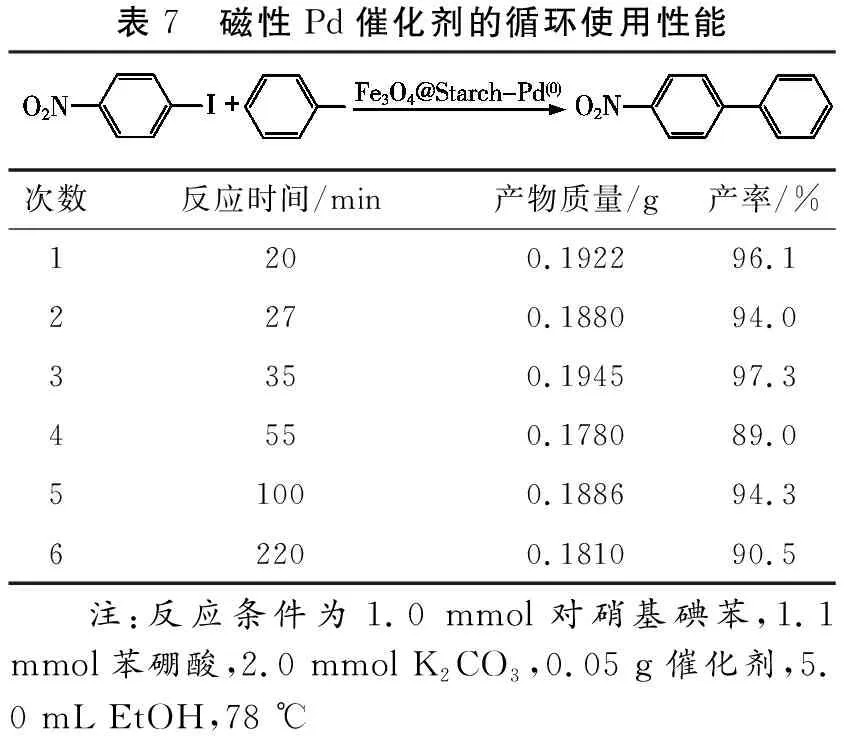
表7 磁性Pd催化劑的循環使用性能次數反應時間/min產物質量/g產率/%1200.192296.12270.188094.03350.194597.34550.178089.051000.188694.362200.181090.5 注:反應條件為1.0 mmol對硝基碘苯,1.1 mmol苯硼酸,2.0 mmol K2CO3,0.05 g催化劑,5.0 mL EtOH,78 ℃
隨后,對重復使用6次后的催化劑進行FT-IR(圖1,曲線c)、XRD(圖2,曲線c)、TGA(圖3,曲線c)及ICP分析。從FT-IR光譜圖和XRD曲線圖中可知重復使用后的催化劑仍保留著Fe3O4、淀粉及Pd(0)的組分,證明該催化劑具有一定的結構穩定性。ICP分析結果表明回收后的催化劑中Pd的含量為0.30 mmol/g,降低了約21%,平均每次回收使用降低4%左右,這一結果可能是因磁性Pd催化劑在催化偶聯反應過程中堿對淀粉有降解作用,導致淀粉結構受損(解聚),對Pd(0)的穩定下降,從而加劇了Pd(0)活性組分在重復使用4次后的團聚現象[9],導致催化劑重復使用4次后,催化活性明顯降低。總之,Fe3O4@Starch-Pd(0)催化劑具有一定的結構穩定性,呈現出較好的重復使用性能。
3 結論
通過淀粉包覆磁性Fe3O4內核并穩定原位形成Pd(0)的方法,成功制備了可磁性回收的Fe3O4@Starch-Pd(0)催化劑。該催化劑對Suzuki偶聯反應具有較好的催化活性,在優化的條件下,20 min內,聯苯的產率即可達到96.1%。此外,該催化劑同樣具有較好的底物普適性和循環使用性能,可催化溴代和碘代芳烴與系列芳基硼酸進行偶聯反應,且催化劑回收使用4次,催化活性僅有稍微降低,表明該催化劑具有一定的結構穩定性。所獲得的研究結果,對生物質基磁性催化劑的開發具有一定的借鑒意義。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
