
STAR基因缺陷致先天性腎上腺皮質增生一例
2022-07-18 05:31:36趙子辰鄧姍田秦杰
生殖醫學雜志 2022年7期
關鍵詞:功能
趙子辰,鄧姍,田秦杰
(中國醫學科學院 北京協和醫學院 北京協和醫院婦產科,國家婦產疾病臨床醫學研究中心,北京 100730)
類脂性先天性腎上腺皮質增生癥(lipoid congenital adrenal hyperplasia,LCAH)是由編碼類固醇激素合成急性調節蛋白(steroidogenic acute regulatory protein,StAR)基因缺陷導致的常染色體隱性遺傳疾病。在中國,LCAH是一種非常罕見的先天性腎上腺類固醇生成缺陷疾病。典型的LCAH表現為腎上腺合成3種類固醇激素(即糖皮質激素、鹽皮質激素、性激素)均缺乏、促腎上腺皮質激素(adrenocorticotropic hormone,ACTH)分泌增加及腎上腺皮質增生。我們報道1例STAR新發基因缺陷致先天性腎上腺皮質增生的中國患者,旨在提高對本病的認識,減少誤診和漏診。
病例資料
患者,14歲,社會性別為女性,因“無第二性征發育,發現染色體為46,XY 8月”入院。
患者系其母第2胎第1產,孕期無特殊。出生時女性外陰,膚色較深。6個月時因反復感冒發熱伴嘔吐、嗜睡、膚色較深就診,考慮“腎上腺功能不全”。8個月時就診北京協和醫院內分泌科,發現高血鉀、低血鈉,血壓一直正常,診斷Addison病,予激素治療,以后規律服用氫化可的松10 mg tid、氟氫可的松50 μg qd,電解質保持正常。4歲診斷亞臨床甲減,口服雷替斯;6歲因“左腹股溝疝“行手術治療,具體不詳。
12歲時因無乳房發育和無月經初潮就診內分泌科。為進一步發現潛在的遺傳疾病,取患者外周血對腎上腺疾病相關基因(腎上腺panel)進行檢測,發現STAR基因突變(表1),并由Sanger測序驗證了以下兩個基因突變:①NM_000349(STAR):c.64+2T>G;②NM_000349(STAR):c.677T>G(p.Val226Gly)。后者為新發突變,此前未在文獻中報道過。隨后,對其父母的外周血進行Sanger測序,驗證了患者的STAR基因c.64+2T>G變異來自父方,c.677T>G(p.V226G)變異來自母方(圖1)。根據美國醫學遺傳學與基因組學學會(ACMG)分類等級,STARc.677T>G功能預測為“可能致病的”。

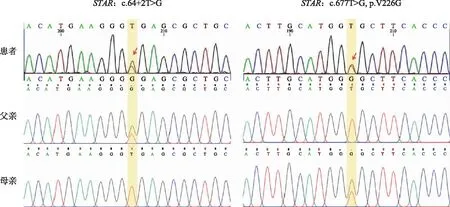
紅色箭頭示患者攜帶的STAR基因突變位點,黃色色條示患者及父母與之對應的基因位點。圖1 患者攜帶的兩個STAR基因變異及家系驗證
13歲時查染色體為46,XY,激素檢查結果如表2所示,促性腺激素、促腎上腺皮質激素升高,雌、孕、雄激素均降低。個人史、家族史無特殊。
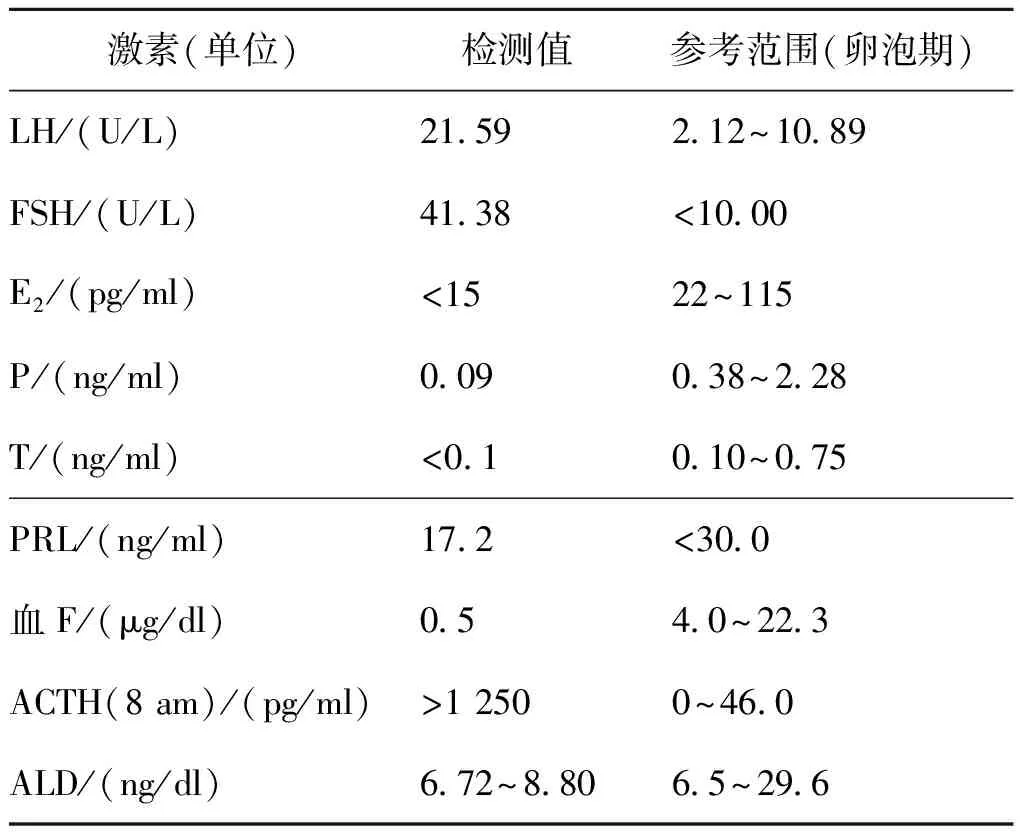
表2 患者13歲時激素水平
入院查體:身高162 cm,體重76 kg,體質量指數(BMI) 28.9 kg/m2。身高年增長2 cm,近半年體重增加4.5 kg。無陰毛、腋毛,膚色偏黑,無喉結。乳房Tanner I期,外陰幼稚女性型,陰蒂不大,大小陰唇發育差,陰道可探入2 cm。
入院后完善全套類固醇激素檢查,顯示類固醇激素大多處于較低水平(表3)。盆腔超聲未見子宮,雙附件區可見中等回聲,右側2.1 cm×1.3 cm、左側2.5 cm×1.4 cm,均未見卵泡。完善腎上腺平掃CT,提示患者雙腎上腺萎縮(圖2)。

表3 患者類固醇激素全套結果
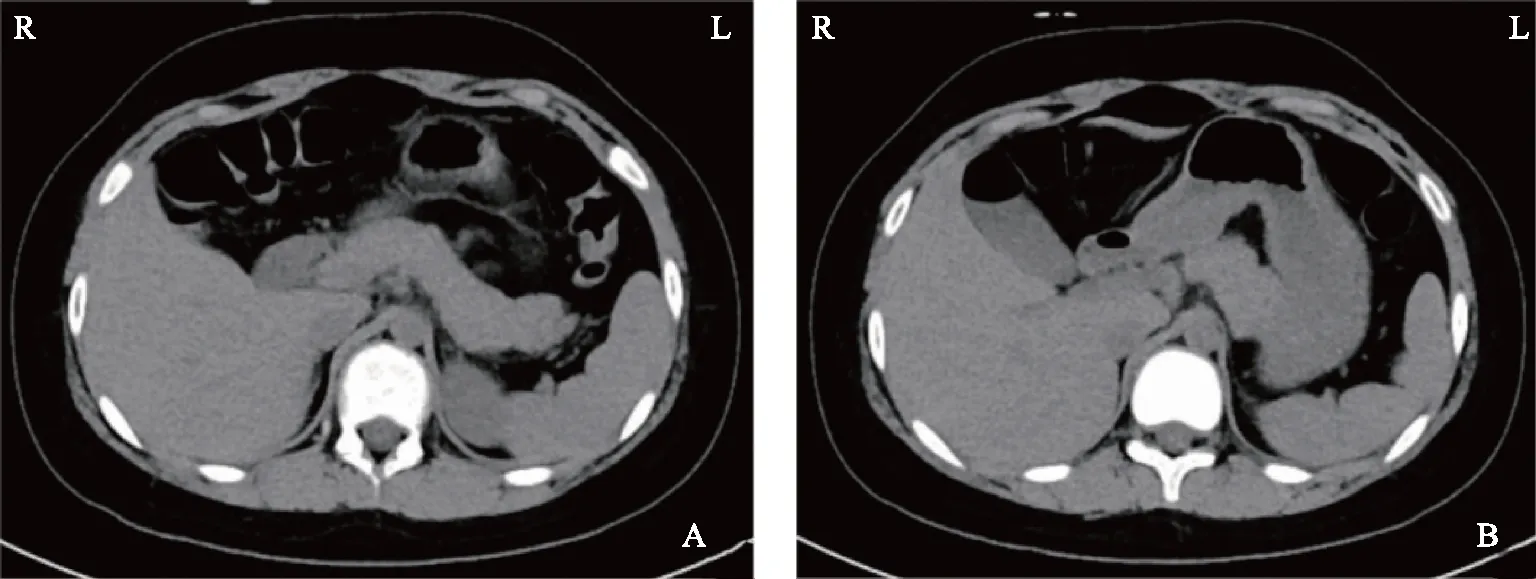
A:截面1;B:截面2。圖2 腎上腺平掃CT
患者嬰兒期出現發熱、嘔吐、嗜睡、低鈉高鉀等腎上腺皮質功能減退癥狀,青春期無自主月經來潮及第二性征發育,基因檢測提示STAR基因缺陷,輔助檢查提示血皮質醇偏低、血ACTH升高。本患者基本符合LCAH的主要表現,家系驗證支持基因缺陷遺傳自父母雙方。不符合之處有兩點:(1)醛固酮水平為正常值低限,并無明顯的降低,但由于患者長期服用氟氫可的松,不除外是外源激素補充后的水平;(2)患者腎上腺CT提示腎上腺萎縮,無明顯增生。
鑒別診斷方面,常見的主要考慮是17α羥化酶缺乏癥,在該疾病中,性激素和糖皮質激素的合成受影響,而鹽皮質激素合成通路是正常的。17α羥化酶缺乏癥的患者具有低水平的皮質醇、性激素和17-羥孕酮,而孕酮、脫氧皮質酮和皮質酮顯著升高,常出現低腎素性高血壓、低血鉀,糖皮質激素不足的癥狀以及性發育異常。本患者孕酮水平很低,且血鉀偏高、無高血壓,同時有明確的STAR基因缺陷,不考慮17α羥化酶缺乏癥。
入院后于全身麻醉下行腹腔鏡探查術。術中見子宮缺如;左側附件:左輸卵管未見,可見發育不全睪丸,2.0 cm×1.2 cm×1.2 cm,其頂端可見脂肪樣組織;右側附件:右輸卵管未見,可見性腺較左側略膨大,3.0 cm×2.0 cm×2.0 cm,其頂端可見脂肪樣組織(圖3);乙狀結腸粘連于側盆壁。分離粘連,切除雙側性腺(圖4)。
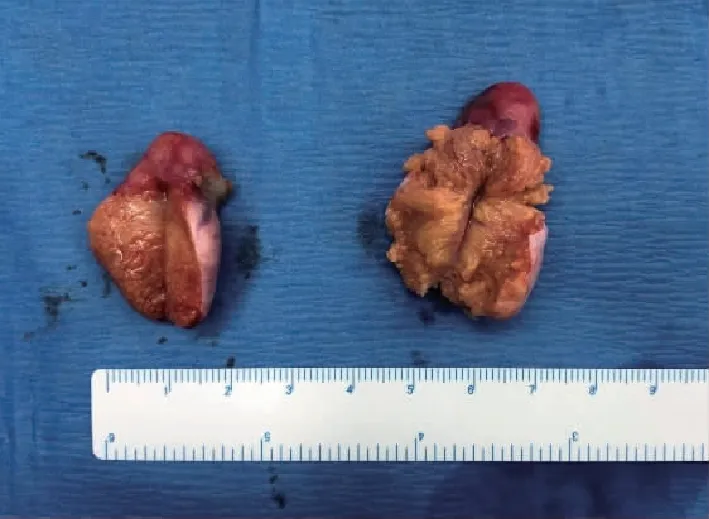
圖4 切除的性腺剖面
術后病理診斷示雙側性腺可見曲細精管及附睪組織,未見生精現象。原發病方面,術后予口服激素治療:氫化可的松tid(劑量每次分別為15 mg、10 mg、10 mg),氟氫可的松75 μg qd,補佳樂1 mg bid。亞臨床甲減方面,繼續予雷替斯口服75 μg qd。
患者術后兩周隨訪,腹部切口愈合良好,繼續服用氫化可的松tid(15 mg、10 mg、10 mg)、氟氫可的松75 μg qd、補佳樂1 mg bid,亞臨床甲減癥狀給予口服雷替斯75 μg qd。術后2.5月隨訪,恢復良好,不易乏力、感冒,但偶爾有膝關節疼痛、腰痛,自9月初自覺乳房稍有發育,睡眠、食欲好,大小便正常。
討 論
類固醇激素合成急性調節蛋白(steroidogenic acute regulatory protein,StAR)由STAR基因編碼,它的作用是介導膽固醇從線粒體外膜向內膜的轉移,這一過程為類固醇激素合成的第一步,為糖皮質激素、鹽皮質激素和性激素的合成提供了底物。
STAR基因缺陷導致上述3種類固醇激素的合成不足,表現為早發的腎上腺功能不全和性腺功能衰竭。性腺發育不良在嚴重程度和發病時間上存在性別差異,46,XY男性患兒在胎兒期已受累,妊娠6~12周期間Leydig細胞不能合成睪酮導致男性胎兒女性化外陰,而睪丸Sertoli細胞不進行類固醇合成,其正常分泌的抗苗勒管激素抑制子宮發育,故患兒無子宮,大多數受影響的46,XY患者由于雄激素不足而缺乏男性化表現。而46,XX患者幾乎均可有正常的青春期發育,在適當的年齡有月經初潮,并有可能生育后代。這可能與體內存在StAR非依賴機制的低水平甾體合成有關。46,XX胎兒卵巢不產生類固醇激素,直到青春期前卵泡細胞并未受刺激或破壞,青春期開始后LH刺激低水平的StAR非依賴性的類固醇合成,每月有一個卵泡募集并受促性腺激素刺激,導致募集細胞內膽固醇累積,細胞受膽固醇酯及其代謝物的毒性作用導致無隨后的孕激素高峰發生,因此患者不排卵,表現為不孕癥;而未募集的卵泡不受影響,每月一個新的募集卵泡可分泌雌激素,維持像正常月經似的周期性子宮雌激素撤退出血;患者可表現有進行性的高促性腺激素性性腺功能減退,一直到卵泡細胞被脂肪堆積而導致功能喪失為止[1]。
本例患者攜帶STAR基因的復合雜合突變,分別位于1號內含子與6號外顯子上。1號內含子中T→G堿基的改變(c.64+2T>G)是一種剪接突變,造成轉錄過程中RNA前體的剪接方式改變,使產生的成熟RNA中含有內含子,造成性狀的改變,是致病性的。既往文獻中曾報道過兩例攜帶這種純合突變的患者[2]。6號外顯子上T→G的點突變(c.677T>G)為錯義突變,導致編碼纈氨酸的密碼子變成編碼甘氨酸的密碼子,從而使多肽鏈中的氨基酸序列發生改變,該突變類型此前未報道過。經家系驗證,該患者的兩個雜合變異分別來自父親和母親,位于不同的基因拷貝上,根據ACMG遺傳變異分類標準預測這一新發突變(STARc.677T>G)是“可能致病的(likely pathogenic)”。ClinVar數據庫中此前記錄有這一位點T堿基缺失導致的移碼突變(c.677del),功能預測同樣是“可能致病的(likely pathogenic)”。
由STAR基因缺陷導致的LCAH疾病表現是兩個獨立事件的結果:STAR基因突變導致的類固醇生成缺陷是主導原因,隨后腎上腺皮質中累積的膽固醇酯或進一步造成激素合成細胞的損傷。最終臨床表型為嬰兒期即出現的腎上腺皮質功能不全的臨床癥狀,如高鉀血癥、低鈉血癥、低容量、酸中毒等,嚴重者在嬰兒期死亡。患者通過鹽皮質激素、糖皮質激素替代治療可以生存到成年。皮質醇缺乏還會引起ACTH的前體阿黑皮素原的生成增加,裂解成ACTH、促黑素細胞激素,間接導致黑色素合成增加,引起色素沉著過度,因此患者的皮膚通常較黑。
大多數LCAH患者表現為雙側腎上腺增生[3]。也有個別文獻報道此類患者的腎上腺是萎縮的,其中有一例中國患者的報道,患者在嬰兒期就表現為早發的腎上腺危象,血皮質醇下降、ACTH升高,女性外陰而染色體為46,XY,符合經典的LCAH,唯有腎上腺是偏小的[4],與本病例類似。國外也有1例類似的腎上腺萎縮的報道,左側腎上腺未見,右側腎上腺縮小[5]。腎上腺萎縮的機制有待進一步研究。另有文獻中報道,非經典型LCAH患者也可有腎上腺縮小的表現[6]。所謂非經典型LCAH,雖有原發性腎上腺功能不全,但性腺功能正常,同時雙側腎上腺無增生。
LCAH在中國是一種非常罕見的先天性腎上腺類固醇生成缺陷,其中完全型LCAH是最嚴重的類型。而在日本和韓國,這種缺乏的患病率在所有先天性腎上腺皮質增生癥(CAH)類型中僅次于21-羥化酶缺乏癥,男/女性別的比例是3∶1,許多受累的個體在嬰兒期死亡(約有1/3靠替代治療存活)[7]。對于存在腎上腺皮質功能減退的任何癥狀或體征、且外生殖器性別不清或呈女性的新生兒,應考慮該診斷;若找不到腎上腺或性腺類固醇生物合成活性的證據,則可確診。
利益沖突聲明所有作者均聲明本研究不存在利益沖突。
猜你喜歡
鐘表(2023年5期)2023-10-27 04:20:44
中華詩詞(2022年6期)2022-12-31 06:41:24
當代陜西(2021年21期)2022-01-19 02:00:26
中學生數理化(高中版.高考數學)(2020年1期)2020-02-20 13:23:44
經濟技術協作信息(2018年11期)2019-01-14 03:07:20
中國科技論壇(2017年7期)2017-07-25 08:49:53
制造技術與機床(2017年3期)2017-06-23 08:11:33
媽媽寶寶(2017年2期)2017-02-21 01:21:24
國際漢語學報(2016年1期)2017-01-20 08:21:20
中國中醫藥現代遠程教育(2014年22期)2014-03-01 04:32:55
