超高效液相色譜-串聯(lián)質(zhì)譜法測(cè)定茶葉中三種農(nóng)藥殘留
2022-08-04 04:17:02劉偉悅
食品工業(yè) 2022年7期
關(guān)鍵詞:標(biāo)準(zhǔn)檢測(cè)
劉偉悅
北京張一元金橋茶葉有限公司(北京 101102)
我國(guó)是茶樹(shù)原產(chǎn)地,茶葉生產(chǎn)歷史悠久。茶葉作為一種健康飲品,深受全世界消費(fèi)者的喜愛(ài)。茶葉中不僅富含茶多酚及兒茶素,還含有鉀、鈣、鎂、錳等多種礦物質(zhì)。茶葉也是我國(guó)重要的經(jīng)濟(jì)作物和出口農(nóng)產(chǎn)品。作為我國(guó)茶葉的重要進(jìn)口國(guó),歐洲和日本制定了極為嚴(yán)苛的農(nóng)藥殘留限量標(biāo)準(zhǔn)。歐盟標(biāo)準(zhǔn)和日本肯定列表中關(guān)于茶葉的農(nóng)殘項(xiàng)目分別為480種[1-2]和276種,農(nóng)殘限量在0.01~30.0 mg/kg之間,國(guó)際食品法典委員會(huì)農(nóng)殘限量(CAC)在0.01~50.0 mg/kg之間,對(duì)于沒(méi)有明確規(guī)定農(nóng)藥殘留限量的農(nóng)藥一律采用不得大于0.01 mg/kg的標(biāo)準(zhǔn)。目前,我國(guó)最新版國(guó)家強(qiáng)制性標(biāo)準(zhǔn)GB 2763—2021《食品安全國(guó)家標(biāo)準(zhǔn)食品中農(nóng)藥最大殘留限量》[3]中涉及茶葉中農(nóng)藥最大殘留限量已經(jīng)達(dá)到106種,農(nóng)藥殘留限量在0.01~50.0 mg/kg之間,對(duì)未涉及農(nóng)藥殘留限量的農(nóng)藥沒(méi)有具體規(guī)定。
為了提高茶葉產(chǎn)量,農(nóng)藥在茶葉病蟲(chóng)防治中得到了廣泛使用,敵敵畏、樂(lè)果和喹硫磷均為有機(jī)磷類農(nóng)藥,廣泛應(yīng)用于茶葉中的殺蟲(chóng)、殺螨劑,由此帶來(lái)的農(nóng)藥殘留可能危害消費(fèi)者健康。隨著生活水平日益提高,人們對(duì)茶葉的質(zhì)量和安全性都有了更高的要求[4]。農(nóng)藥殘留分析作為一項(xiàng)對(duì)復(fù)雜混合物中痕量組分的分析技術(shù)。目前,國(guó)內(nèi)外農(nóng)藥殘留的檢測(cè)方法主要有氣相色譜法、液相色譜法、氣相色譜-質(zhì)譜法、氣相色譜-串聯(lián)質(zhì)譜法、液相色譜-串聯(lián)質(zhì)譜法等[5-6]。其中,液相色譜-質(zhì)譜聯(lián)用法和氣相色譜-質(zhì)譜聯(lián)用法利用被測(cè)樣品離子質(zhì)荷比,定性定量更加準(zhǔn)確,檢測(cè)靈敏度更高,對(duì)于茶葉類復(fù)雜基質(zhì)中農(nóng)藥殘留檢測(cè)有其獨(dú)特的技術(shù)優(yōu)勢(shì)。
研究建立了適用于檢測(cè)茶葉中農(nóng)藥殘留的超高效液相色譜-串聯(lián)質(zhì)譜法,該方法簡(jiǎn)單快速、分離度好、重現(xiàn)性好、精密度高,可滿足同時(shí)檢測(cè)茶葉中敵敵畏、樂(lè)果和喹硫磷3種農(nóng)藥殘留的要求[7-13]。
1 材料與方法
1.1 材料與儀器
丙酮中敵敵畏、樂(lè)果、喹硫磷質(zhì)量濃度均為1 000 μg/mL(農(nóng)業(yè)部環(huán)境保護(hù)科研監(jiān)測(cè)所);乙腈、丙酮、甲醇、甲苯、甲酸[色譜純,賽默飛世爾科技(中國(guó))有限公司];純凈水(杭州娃哈哈有限公司);無(wú)水硫酸鈉(分析純,北京化工廠),經(jīng)550 ℃灼燒4 h,儲(chǔ)存于干燥器中,冷卻后備用;Cleanert TPT固相萃取柱(Agela公司);0.22 μm尼龍微孔過(guò)濾膜(津騰公司)。
UPLC/TSQ Quantum U1tra超高效液相色譜-三重四級(jí)桿質(zhì)譜聯(lián)用儀[配有電噴霧離子源(ESI)及Xcalibur工作站軟件,賽默飛世爾科技(中國(guó))有限公司];Hypersil GOLD C8色譜柱[100 mm×2.1 mm,1.9 μm,賽默飛世爾科技(中國(guó))有限公司];RE-2000B旋轉(zhuǎn)蒸發(fā)器(上海亞榮生化儀器廠);QL-901渦旋混合器(海門(mén)市其林貝爾儀器制造有限公司);XHF-DY高速分散器(寧波新芝生物科技股份有限公司);KH-100DB型數(shù)控超聲波清洗器(昆山禾創(chuàng)超聲儀器有限公司);SE202F電子分析天平(奧豪斯);TDL-40B臺(tái)式低速離心機(jī)(飛鴿牌);HY-5A型回旋式振蕩器(江蘇省金壇市榮華儀器制造有限公司)。
1.2 試驗(yàn)方法
1.2.1 待測(cè)液制備
稱取粉碎均勻的試樣5 g(精確至0.01 g),用20.0 mL的乙腈振蕩提取1 h,在4 000 r/min下離心5 min。取4 mL上清液,過(guò)Cleanert TPT固相萃取柱(萃取柱中加入約2 cm高無(wú)水硫酸鈉,用10 mL乙腈+甲苯(3+1,V/V)洗脫液預(yù)洗,棄去流出液)凈化,用20 mL乙腈+甲苯(3+1,V/V)洗脫,收集洗脫液,40 ℃條件下水浴,旋蒸至近干,乙腈+水(3+2,V/V)定容1 mL,過(guò)0.22 μm微孔過(guò)濾膜,混勻,待UPLC-MS/MS測(cè)定,以基質(zhì)標(biāo)準(zhǔn)溶液工作曲線定量。
1.2.2 標(biāo)準(zhǔn)曲線的繪制
混合標(biāo)準(zhǔn)儲(chǔ)備液的配制:將1 000 μg/mL丙酮中敵敵畏、樂(lè)果、喹硫磷農(nóng)藥標(biāo)準(zhǔn)溶液稀釋為100 μg/mL標(biāo)準(zhǔn)儲(chǔ)備液于-20 ℃保存。分別吸取樂(lè)果和喹硫磷標(biāo)準(zhǔn)儲(chǔ)備液100 μL加入10.0 mL容量瓶中,用丙酮定容至刻度,得到1 μg/mL的混合標(biāo)準(zhǔn)工作溶液A,同樣方法得到1 μg/mL的敵敵畏、樂(lè)果、喹硫磷混合標(biāo)準(zhǔn)工作溶液B,于-20 ℃保存。
基質(zhì)標(biāo)準(zhǔn)曲線的制備:同1.2.1小節(jié)待測(cè)液制備的方法,用未檢出待測(cè)農(nóng)藥的綠茶樣品制作基質(zhì)空白溶液,分取16 mL上清液過(guò)柱,20 mL乙腈+甲苯(3+1)洗脫,用乙腈+水(3+2)定容至8 mL,過(guò)0.22 μm濾膜,所得溶液為基質(zhì)空白溶液。準(zhǔn)確配制基質(zhì)空白中3種農(nóng)藥的質(zhì)量濃度為10,20,50,100,200,400和600 μg/L進(jìn)行測(cè)定。
1.2.3 液相色譜條件
Hypersil GOLD C8色譜柱(100 mm×2.1 mm,1.9 μm)。流動(dòng)相組成:流動(dòng)相A為0.1%甲酸,流動(dòng)相B為乙腈。流動(dòng)相梯度洗脫程序見(jiàn)表1;色譜柱溫度40oC,進(jìn)樣量為5 μL。
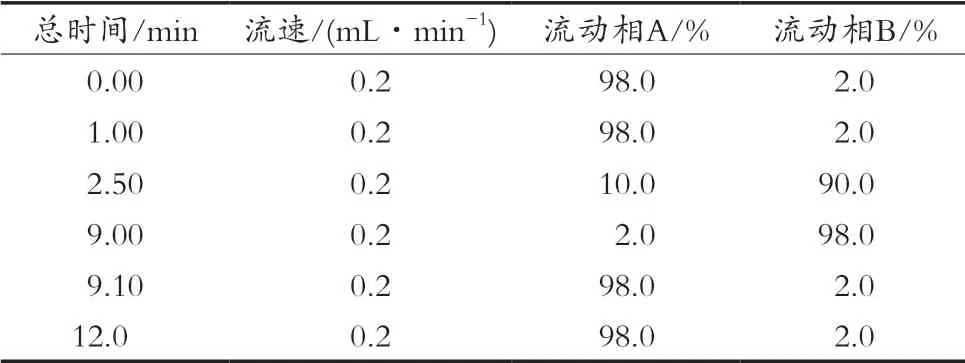
表1 流動(dòng)相梯度洗脫程序
1.2.4 質(zhì)譜條件
電離方式ESI+;噴霧電壓:3 200 V;離子傳輸管溫度:320 ℃;蒸發(fā)溫度300 ℃;鞘氣:氮?dú)猓髁?30 L/h;輔助氣:氮?dú)猓髁?80 L/h;碰撞氣:氬氣,碰撞氣壓力1.5 mTorr;氮?dú)夂蜌鍤饧兌染笥?9.999%;選擇離子多反應(yīng)監(jiān)測(cè)模式掃描(SRM)。
2 結(jié)果與分析
2.1 色譜-質(zhì)譜條件的優(yōu)化
在優(yōu)化好的色譜條件下,采用全掃描Full Scan方式對(duì)單一標(biāo)準(zhǔn)溶液2 μg/mL進(jìn)樣,獲得化合物的特征離子,選擇離子豐度最高的[M+H]+m/z作為母離子;然后進(jìn)行產(chǎn)物離子掃描,找到特征離子及其最佳CE。試驗(yàn)選擇每種化合物豐度較高的兩個(gè)離子分別作為定量離子和定性離子。化合物英文名稱、保留時(shí)間及質(zhì)譜參數(shù)見(jiàn)表2。
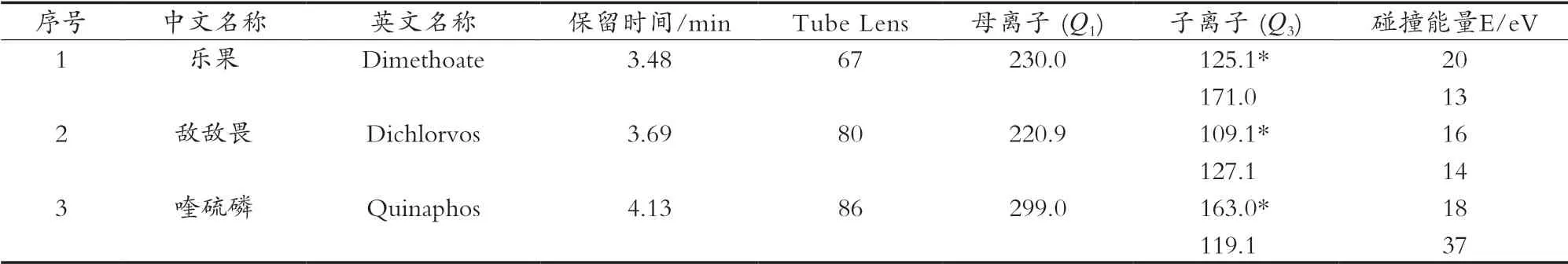
表2 保留時(shí)間和質(zhì)譜參數(shù)
2.2 提取過(guò)程的優(yōu)化
2.2.1 提取溶劑的優(yōu)化
在農(nóng)藥殘留提取的過(guò)程中,根據(jù)農(nóng)藥的極性、性質(zhì)等因素選擇合適的提取溶劑。在農(nóng)藥殘留檢測(cè)的過(guò)程中,常見(jiàn)的提取溶劑有乙腈、丙酮、甲醇、正己烷、石油醚等,研究以回收率為參考指標(biāo),回收試驗(yàn)添加質(zhì)量分?jǐn)?shù)水平為0.1 mg/kg,用乙腈、丙酮和甲醇3種有機(jī)溶劑作為提取溶劑分別做3平行試驗(yàn),回收率見(jiàn)圖1。
從圖1可以看出,甲醇作為提取劑時(shí)樂(lè)果和喹硫磷回收率極低,敵敵畏回收率為零。由于甲醇的溶解性較好,在提取的過(guò)程中,提取液比較渾濁,最后定容,待上機(jī)的溶液會(huì)分層,這可能是導(dǎo)致三種農(nóng)殘回收率都很低的原因。乙腈和丙酮提取的回收率比較接近,但是乙腈對(duì)于色素和油脂的提取能力不強(qiáng),因此可以減小雜質(zhì)的干擾,提取的回收率整體更優(yōu)于丙酮,因此選擇乙腈作為提取溶劑。
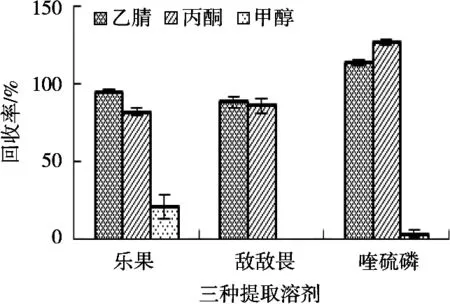
圖1 三種提取溶劑農(nóng)殘回收率對(duì)比(n=3)
2.2.2 提取方式的優(yōu)化
研究以回收率為參考指標(biāo),回收試驗(yàn)添加質(zhì)量分?jǐn)?shù)水平為0.1 mg/kg,用超聲、振蕩和均質(zhì)三種方式提取,分別做3組平行試驗(yàn),回收率見(jiàn)圖2。
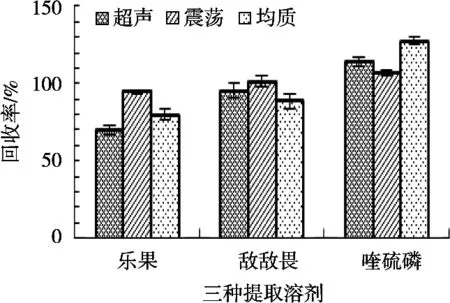
圖2 三種提取方式農(nóng)殘回收率對(duì)比(n=3)
從圖2可以看出,超聲提取方式對(duì)樂(lè)果的回收率相對(duì)較低,均質(zhì)提取方式對(duì)喹硫磷的回收率相對(duì)較高,這可能是超聲和均質(zhì)提取方式使一部分干擾物被提取出來(lái),增強(qiáng)了茶葉的基質(zhì)干擾。而通過(guò)振蕩提取的方式,三種農(nóng)藥的回收率表現(xiàn)良好。因此,試驗(yàn)選擇振蕩提取。
2.2.3 洗脫液提體積的優(yōu)化
研究以回收率為參考指標(biāo),回收試驗(yàn)添加質(zhì)量分?jǐn)?shù)水平為0.1 mg/kg,用乙腈+甲苯(3+1,V/V)作洗脫液,Cleanert TPT固相萃取柱進(jìn)行凈化,洗脫液體積分別為10,15,20,25和30 mL,分別做3組平行試驗(yàn),回收率見(jiàn)圖3。
從圖3可以看出,喹硫磷的回收率受洗脫液體積影響較小。對(duì)于樂(lè)果和敵敵畏,隨著洗脫液體積的增加,其平均回收率總體呈上升趨勢(shì);但超過(guò)20 mL之后,隨著洗脫液體積的增加,其回收率變化不明顯。基于添加回收率要求和試驗(yàn)操作的實(shí)際需求,選擇20 mL乙腈+甲苯(3+1,V/V)作為洗脫液。
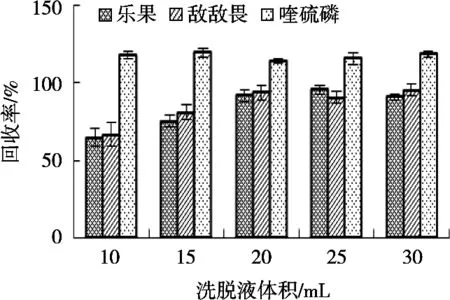
圖3 洗脫液體積對(duì)農(nóng)殘回收率的影響(n=3)
2.3 標(biāo)準(zhǔn)曲線與線性范圍
按上述方法配制標(biāo)準(zhǔn)曲線,以目標(biāo)組分的響應(yīng)面積(y)和對(duì)相應(yīng)的質(zhì)量濃度(x,μg/L)繪制標(biāo)準(zhǔn)曲線。結(jié)果發(fā)現(xiàn),三種農(nóng)藥的質(zhì)量濃度在10~600 μg/L范圍內(nèi)具有良好的線性關(guān)系,相關(guān)系數(shù)均大于0.99。以10倍信噪比(S/N)計(jì)算方法的定量限[14-20],茶葉中三種農(nóng)藥線性回歸方程、相關(guān)系數(shù)、定量限和限值見(jiàn)表3。
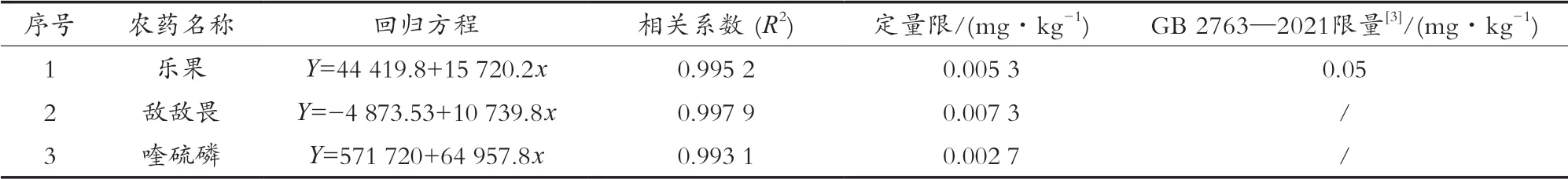
表3 茶葉中3種農(nóng)藥線性回歸方程、相關(guān)系數(shù)、定量限和限值
2.4 加標(biāo)回收和精密度
選擇空白樣品進(jìn)行加標(biāo)處理,處理方法同待測(cè)液的制備方法。按照所建立的方法處理、測(cè)定,加標(biāo)水平為0.01,0.02,0.1和0.4 mg/kg,每個(gè)加標(biāo)量重復(fù)6次,驗(yàn)證方法的精密度。空白樣品中3種農(nóng)藥加標(biāo)回收結(jié)果見(jiàn)表4。從表4中可以看出,3種農(nóng)藥的加標(biāo)回收率在68.7%~114.9%之間,精密度為2.7%~10.8%。回收率和精密度均符合農(nóng)藥殘留檢測(cè)要求。
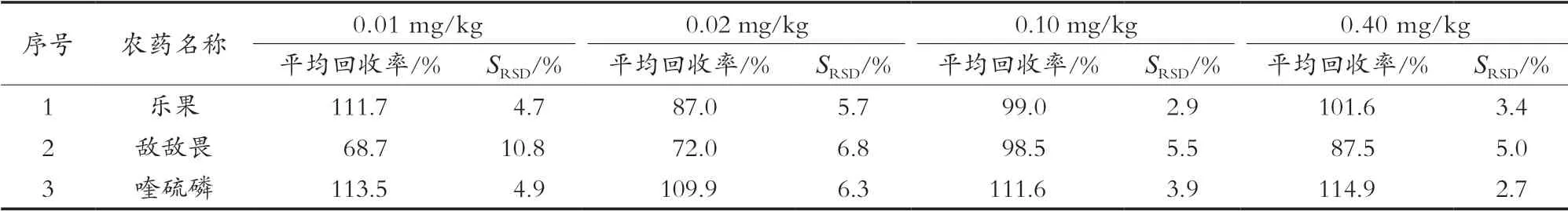
表4 農(nóng)藥加標(biāo)回收率及精密度
2.5 5種茶葉樣品加標(biāo)回收率分析
對(duì)綠茶、紅茶、茉莉花茶、大紅袍和普洱生茶5種茶葉進(jìn)行3種農(nóng)殘加標(biāo)試驗(yàn)。制備均勻樣品,采用此方法提取、凈化、上機(jī)測(cè)試,添加水平及回收率見(jiàn)表5。從表5中可以看出,5種茶葉中3種農(nóng)藥的回收率在66.6%~113.6%之間,表明此方法可用于茶葉中3種農(nóng)藥殘留的同時(shí)檢測(cè)。

表5 5種茶葉加標(biāo)回收
2.6 樣品測(cè)定
隨機(jī)抽取綠茶、紅茶、黑茶、烏龍茶、白茶、茉莉花茶等六種茶樣,共198批次,采用上述建立的檢測(cè)方法對(duì)樣品進(jìn)行檢測(cè)。結(jié)果表明,敵敵畏、樂(lè)果和喹硫磷均未被檢出。
3 結(jié)論
用乙腈振蕩提取1 h,Cleanert TPT固相萃取柱凈化,超高效液相色譜-串聯(lián)質(zhì)譜法檢測(cè),建立了茶葉中同時(shí)測(cè)定樂(lè)果、敵敵畏和喹硫磷殘留量的測(cè)定方法,且方法線性相關(guān)性好、簡(jiǎn)單快捷、靈敏度高,在回收率、定量限和精密度等方面均能滿足我國(guó)、歐盟和日本等國(guó)家對(duì)于茶葉中農(nóng)藥殘留量的檢測(cè)要求。研究一方面可為茶葉產(chǎn)品標(biāo)準(zhǔn)的完善提供數(shù)據(jù),另一方面可為監(jiān)管部門(mén)對(duì)茶葉產(chǎn)品的風(fēng)險(xiǎn)監(jiān)測(cè)提供方法參考。
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2020年12期)2021-01-18 06:57:46
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2020年12期)2021-01-18 06:57:46
當(dāng)代陜西(2019年8期)2019-05-09 02:22:48
動(dòng)漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
家庭影院技術(shù)(2018年4期)2018-05-09 07:07:52
海峽科技與產(chǎn)業(yè)(2016年3期)2016-05-17 04:32:12

- 食品工業(yè)的其它文章
- 山東省食品科學(xué)技術(shù)學(xué)會(huì)團(tuán)體標(biāo)準(zhǔn)立項(xiàng)評(píng)審暨專家研討會(huì)召開(kāi)
- 鷹潭市食安辦發(fā)布關(guān)于預(yù)防夏季野生蘑菇中毒的溫馨提示
- 嘉峪關(guān)市市場(chǎng)監(jiān)管局鏡鐵分局開(kāi)展保健食品專項(xiàng)檢查
- 【“守查保”專項(xiàng)行動(dòng) 】滕州市突出抓好進(jìn)口冷鏈?zhǔn)称繁O(jiān)管各項(xiàng)工作
- 孝感市2項(xiàng)地理標(biāo)志產(chǎn)品省級(jí)地方標(biāo)準(zhǔn)發(fā)布
- 宜春市召開(kāi)食品(保健食品)生產(chǎn)企業(yè)食品安全責(zé)任約談會(huì)