不同產(chǎn)地的當歸中苯酞類成分的含量測定及聚類分析
2022-08-24 08:31:28周德勇拱健婷宋京美胡銀燕
中國醫(yī)藥導(dǎo)報 2022年19期
龔 韜 楊 薇 王 夏 周德勇 拱健婷 李 莉 宋京美 胡銀燕
北京市臨床藥學研究所,北京 100035
當歸是傘形科植物當歸的干燥根,性甘、辛、溫;歸肝、心、脾經(jīng)。具有補血活血、調(diào)經(jīng)止痛,潤腸通便之功效[1]。主要分布在甘肅、云南、四川等地。現(xiàn)代研究表明,當歸中的化合物主要包括揮發(fā)油、有機酸、多糖和黃酮等成分,其中揮發(fā)油為當歸的有效成分之一,具有改善學習記憶能力、抗氧化損傷等多重藥理作用,按化學結(jié)構(gòu)其主要成分可分為苯酞類和苯酞二聚體類化合物,其中藁本內(nèi)酯的含量最高[2-9]。2020 年版《中華人民共和國藥典》[1]中當歸含量測定項下僅收載了揮發(fā)油總量和阿魏酸含量,但對揮發(fā)油中的具體成分并未規(guī)定其含量測定項。基于此,為更系統(tǒng)、更全面地進行當歸的質(zhì)量評價,本研究建立了多波長高效液相色譜法(high performance liquid chromatograph,HPLC)測定當歸中洋川芎內(nèi)酯I、洋川芎內(nèi)酯A、藁本內(nèi)酯和丁烯基苯酞4 種苯酞類成分的含量,并對不同產(chǎn)地當歸的頭和身兩個部位的洋川芎內(nèi)酯I、藁本內(nèi)酯和丁烯基苯酞3 種成分進行聚類分析,以期為當歸的質(zhì)量評價、標準完善以及合理的開發(fā)利用提供參考和依據(jù)。
1 儀器與材料
1.1 儀器
Thermo UltiMate3000 型超高效液相色譜儀(美國賽默飛世爾科技公司);Mettler Toledo XP105 型十萬分之一電子天平(瑞士梅特勒托利多公司)。
1.2 材料
對照品:洋川芎內(nèi)酯I(貨號:P10OBF45372)、洋川芎內(nèi)酯A(貨號:P03N8F47409)、丁烯基苯酞(貨號:T04N8Z47410)均購自上海源葉生物科技有限公司;藁本內(nèi)酯(貨號:C10106732)購自上海麥克林生化科技有限公司。乙腈為色譜純;其余試劑為分析純。不同產(chǎn)地當歸樣品來源信息見表1,經(jīng)北京市臨床藥學研究所中藥資源室李莉研究員鑒定為傘形科植物當歸Diels 的干燥根。
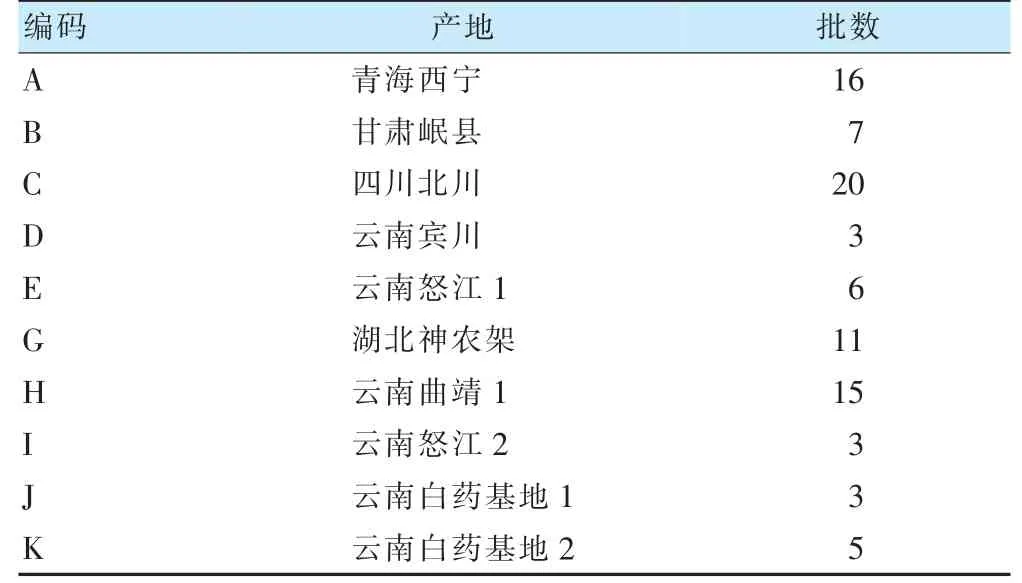
表1 不同產(chǎn)地當歸樣品來源信息
2 方法與結(jié)果
2.1 對照品溶液的制備
取4 種對照品適量,精密稱定,置棕色量瓶中,加甲醇制成每1 ml 含洋川芎內(nèi)酯I 2.34 μg、洋川芎內(nèi)酯A 4.46 μg、藁本內(nèi)酯166.4 μg、丁烯基苯酞2.08 μg的混合溶液,即得。
2.2 供試品溶液的制備
取當歸粉末(B-2-頭)(過三號篩)約0.3 g,精密稱定,置具塞錐形瓶中,精密加入甲醇25 ml,密塞,稱定重量,超聲處理30 min,放冷,再稱定重量,用甲醇補足減失的重量,搖勻,靜置,取上清液濾過,取續(xù)濾液,即得。
2.3 色譜條件
以十八烷基硅烷鍵合硅膠柱(Agilent Eclipse Plus C18,4.6 mm×150 mm,3.5 μm)為色譜柱;流動相為乙腈(A)-水(B)梯度洗脫(0~20 min,92%→74%B;20~25 min,74%→60%B;25~60 min,60%B);柱溫35℃;檢測波長:洋川芎內(nèi)酯I、洋川芎內(nèi)酯A 為284 nm,藁本內(nèi)酯為326 nm,丁烯基苯酞為260 nm;流速1 ml/min。此條件下4 種成分與其他組分均能達到基線分離,色譜圖見圖1。
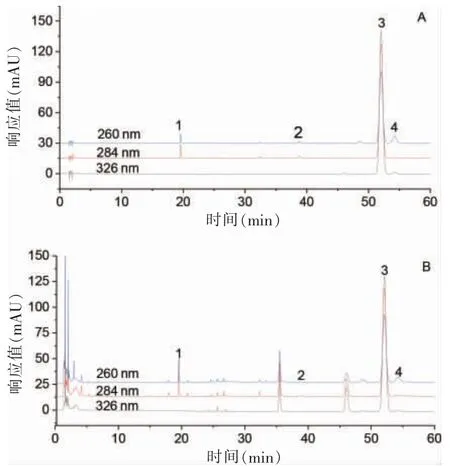
圖1 當歸4 種苯酞類成分高效液相色譜圖譜
2.4 線性關(guān)系考察
取洋川芎內(nèi)酯I、洋川芎內(nèi)酯A、藁本內(nèi)酯、丁烯基苯酞適量,精密稱定,置棕色量瓶中,加甲醇使溶解并稀釋至刻度,搖勻,制成一系列混合對照品溶液,濃度分別為5.85×10-4~2.34×10-2、1.12×10-3~4.46×10-2、4.16×10-2~1.664、5.20×10-4~2.08×10-2mg/ml。按“2.3”項下色譜條件分別進樣10 μl,以對照品進樣量為橫坐標,以峰面積為縱坐標,繪制標準曲線,計算回歸方程。見表2。

表2 4 種苯酞類成分的線性關(guān)系
2.5 方法學考察
2.5.1 穩(wěn)定性考察 精密吸取“2.2”項下同一份供試品溶液10 μl,于0、4、8、12、16、20、24 h 按“2.3”項下色譜條件進樣測定,記錄峰面積。計算得洋川芎內(nèi)酯I、洋川芎內(nèi)酯A、藁本內(nèi)酯、丁烯基苯酞4 種成分峰面積的RSD 值分別為4.07%、1.92%、0.68%、4.23%,表明供試品溶液在24 h 內(nèi)穩(wěn)定性較好。
2.5.2 重復(fù)性考察 精密稱取同一批次當歸樣品(B-2-頭)6 份,按“2.2”項下方法平行制備供試品溶液,分別進樣10 μl,按“2.3”項下色譜條件測定,計算得到洋川芎內(nèi)酯I、洋川芎內(nèi)酯A、藁本內(nèi)酯、丁烯基苯酞4 種成分含量分別為0.0346%、0.0181%、1.0410%、0.0203%,RSD 分別為2.86%、3.24%、1.10%、1.59%,表明該方法重復(fù)性良好。
2.5.3 準確度(回收率)考察 采用加樣回收法。精密稱取已知含量的同一批當歸樣品(B-2-頭)0.15 g,共9 份,精密加入已知含量的對照品溶液25 ml,按“2.2”項下方法制備供試品溶液,分別進樣10 μl,按“2.3”項下色譜條件測定,計算含量及加樣回收率。見表3。

表3 當歸四種苯酞類成分加樣回收試驗結(jié)果
2.6 不同產(chǎn)地當歸樣品測定
不同產(chǎn)地的當歸樣品充分干燥后,按當歸頭、當歸身不同部位進行取樣,粉碎,按“2.2”項下方法制備供試品溶液,并按“2.3”項下色譜條件測定,得到洋川芎內(nèi)酯I、洋川芎內(nèi)酯A、藁本內(nèi)酯、丁烯基苯酞4 種成分含量。
2.7 數(shù)據(jù)處理結(jié)果
采用Graphpad Prism 8.0 軟件對10 個產(chǎn)地的89 批樣品做雨點分布圖,結(jié)果見圖2。結(jié)果顯示,不同產(chǎn)地的當歸樣品中4 種苯酞類成分含量相差較大,部分產(chǎn)地樣品中無洋川芎內(nèi)酯A;洋川芎內(nèi)酯I 和丁烯基苯酞的含量數(shù)據(jù)比藁本內(nèi)酯要分散,同一產(chǎn)地的不同批次間數(shù)據(jù)波動較大。
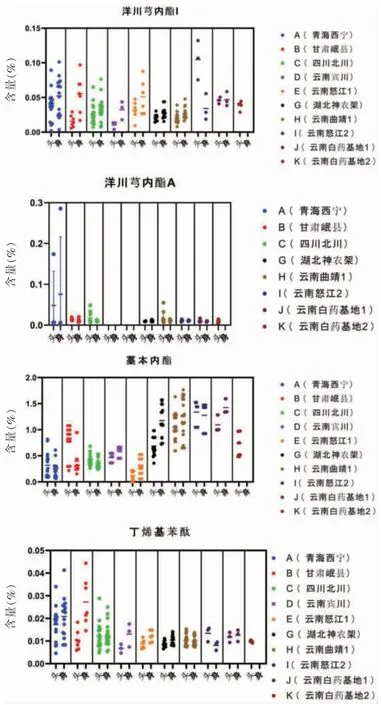
圖2 不同產(chǎn)地當歸中苯酞類成分雨點圖
2.8 聚類分析
以所測洋川芎內(nèi)酯I、藁本內(nèi)酯、丁烯基苯酞3 種成分的含量作為原始數(shù)據(jù),采用SPSS 26.0 軟件標準化處理數(shù)據(jù),以Ward 接法結(jié)合Squared Euclidean Distance 距離計算方法對10 個產(chǎn)地的當歸樣品按不同部位進行聚類分析,結(jié)果見圖3。對于當歸頭,當判定距離<25 時,樣品可聚為2 類,產(chǎn)地編碼H、I、J 為一類,其余產(chǎn)地為一類;當判定距離<5 時,樣品則可聚為4 類,分別是產(chǎn)地編碼B、G、K 為一類,A、C、D 為一類,E 單獨為一類;H、I、J 為一類。對于當歸身,當判定距離<25 時,樣品可聚為2 類,產(chǎn)地編碼G、H、I、J為一類,其余產(chǎn)地的為一類;當判定距離<5 時,樣品則聚為3 類,分別是產(chǎn)地編碼G、H、I、J 為一類,A、B、C、E 為一類,D、K 為一類。
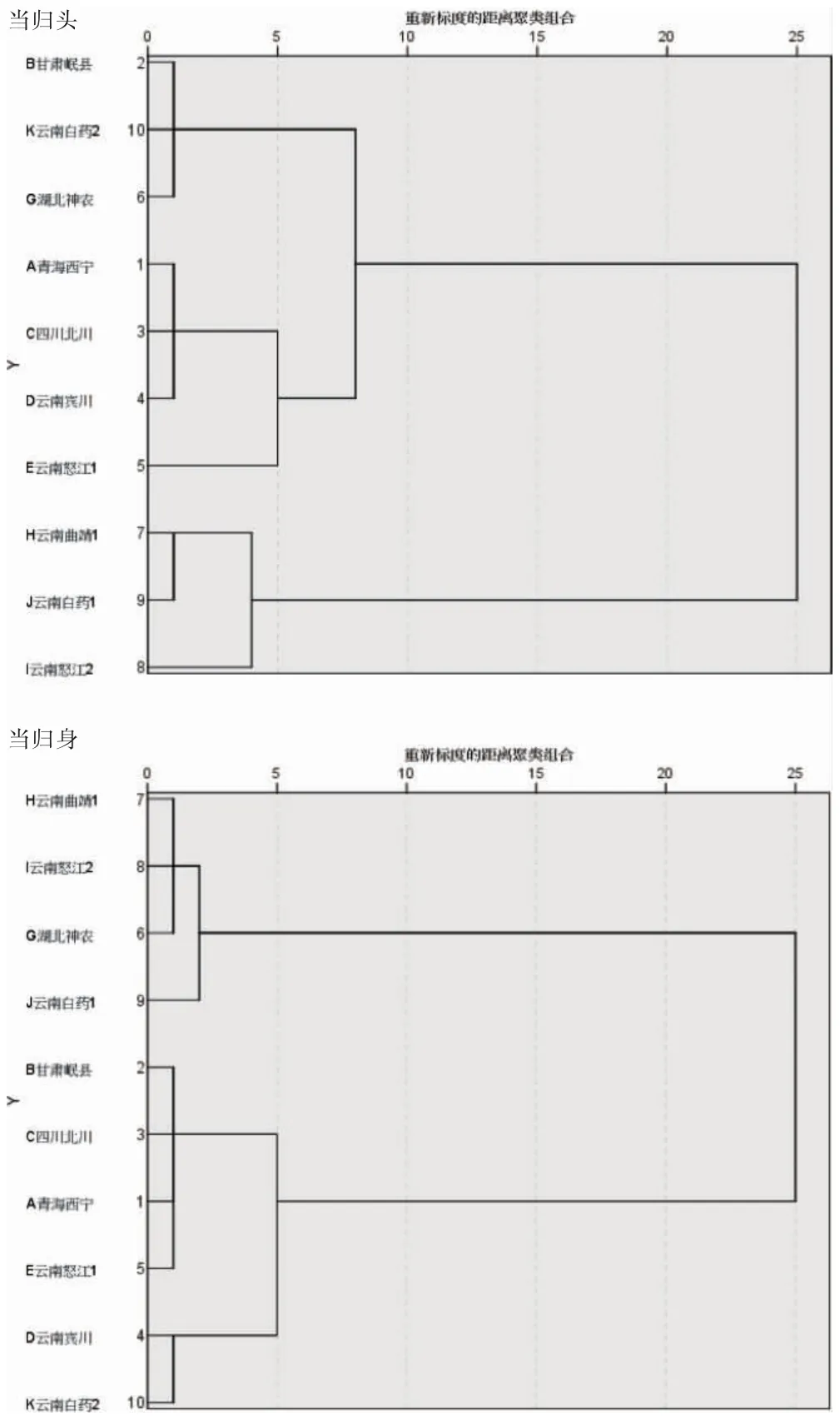
圖3 不同產(chǎn)地當歸的聚類分析
3 討論
3.1 流動相的選擇
參考了已有的關(guān)于當歸有效成分的含量測定及指紋圖譜、特征圖譜的報道[10-20],考察對比了不同色譜系統(tǒng)對樣品分離的影響,最終選用乙腈-水系統(tǒng)作為色譜系統(tǒng),并采用梯度洗脫。
3.2 檢測波長的選擇
由于樣品中4 種成分的含量相差懸殊,藁苯內(nèi)酯的含量是另3 種成分的10~100 倍,因此采用了各個成分的最佳檢測波長,以期收到較好的檢出結(jié)果。
3.3 供試品溶液制備條件的優(yōu)化
由于苯酞類成分穩(wěn)定性差,故選取超聲處理作為提取方式,以甲醇作為提取溶媒。根據(jù)提取溶媒體積和提取時間的考察結(jié)果,確定樣品的提取方式為甲醇超聲處理30 min,溶媒體積為25 ml。
3.4 小結(jié)
本研究建立了當歸中4 種苯酞類成分的含量測定方法,該方法操作簡單,專屬性強,準確性高。不同產(chǎn)地當歸中苯酞類成分的含量差異顯著,可為有關(guān)當歸藥材開發(fā)產(chǎn)地選擇提供基礎(chǔ)研究數(shù)據(jù)。聚類分析根據(jù)不同閾值范圍可把10 個產(chǎn)地89 批當歸樣品分為2 大類或3~4 小類,總體來看,云南曲靖1、云南怒江2 和云南白藥基地1 三個產(chǎn)地的可歸屬為一類,提示以藁本內(nèi)酯等苯酞類化學成分為指標的當歸藥材在地理環(huán)境方面的差異是客觀存在的。本研究由于采集的各個產(chǎn)地的當歸樣品數(shù)量一致性欠佳,樣本數(shù)不齊,可能會造成數(shù)據(jù)統(tǒng)計上的偏差。當歸在臨床上應(yīng)用非常廣泛,因此有必要進一步擴大樣本量以掌握各個產(chǎn)地當歸藥材的總體情況,更全面、系統(tǒng)地進行質(zhì)量評價[21-25]。
