金屬聯吡啶/鄰菲咯啉功能MOFs材料的合成與催化應用
2022-11-10 01:44:12侯寶明譚春霞
上海師范大學學報·自然科學版 2022年5期
關鍵詞:催化劑
侯寶明,譚春霞
(上海師范大學化學與材料科學學院,上海 200234)
0 引言
催化是化學反應非常重要的過程,現代化學品中85%以上是通過催化過程獲得的.因此,開發穩定、高效、可回收的催化劑是化學領域的研究熱點.在這種情況下,多相催化是化學工業中最重要的轉化技術之一,其巨大的潛力為開發新型多相催化劑奠定了基礎.傳統的無機材料,包括沸石、介孔二氧化硅、金屬氧化物和碳材料以及聚合物已被廣泛用作負載型催化劑的載體.然而,由于這些多相催化劑的催化位點隨機、非晶態分布以及活性中心的易失活,導致其催化效率較低.自從出現了新型多孔晶體材料,如金屬有機框架(MOFs)以來,這種情況得以改觀.這些材料具有結晶度高、孔隙率永久、結構多樣、功能可調,以及良好的熱穩定性和化學穩定性等優點,因此它們的骨架和孔可用于設計多種催化劑.
在過去的研究中,通過組合各種有機連接物和含金屬節點的金屬簇,研究人員探索出了數百萬種不同骨架和特定孔隙的結構.這些設計為探索高效多相催化活性的MOFs提供了機會.與傳統的無機材料和有機聚合物相比,MOFs具有多孔性、穩定性、可調節性和可設計性等多種特性,因此具有廣闊的應用前景.由于其結構有序,MOFs允許通過預先設計或后期合成的方法精確固定分子催化劑,以制造具有特定局部微環境的催化活性中心.此外,MOFs的孔隙率可以促進底物和產物的快速轉移,從而加速催化反應.最后,MOFs的非均質性使其不僅易于在反應中分離和回收,而且適合于工業連續流動合成.MOFs基催化劑可以將均相催化劑和非均相催化劑的優點集成到一種材料中.
螯合配體2,2’-聯吡啶(bpy)和1,10-菲咯啉(phen)是剛性平面、疏水和電子共軛的雜環芳香族配體,廣泛應用于配位化學[1]和均相有機催化領域[2].由于其具有氧化還原惰性,易于功能化和與各種金屬離子的良好配位能力,以及較低的制備成本,在精細化學合成中成為了膦基配體的最優替代品[3].此外,由于聯吡啶/鄰菲咯啉(bpy/phen)中的空p*軌道,可以與一些金屬,如銥(Ir(Ⅲ))、釕(Ru(Ⅱ))和錸(Re(Ⅰ))進行配位,形成的配合物在可見光譜中具有很強的金屬-配體電荷轉移(MLCT)特征.此外,從綠色化學和可持續發展的角度來看,用bpy和phen衍生配體分離和回收均相催化劑具有重要意義.本文作者綜述了金屬修飾的聯吡啶/鄰菲咯啉(M(bpy)/M(phen))功能化MOFs催化劑的設計和制備的最新研究進展,并闡述了它們的催化應用(熱催化、光催化和電催化).結論和展望部分還介紹了MOFs基M(bpy)/M(phen)催化劑在高級催化中的機遇和挑戰.
1 M(bpy)/M(phen)功能化MOFs催化劑的設計策略
由于MOFs材料的合成方法和表征技術的進步,MOFs的催化性能可以得到更好的設計和控制.一般來說,MOFs的催化活性主要取決于組分的性質、催化位點的可及性、永久孔隙率和底物可及的納米孔空間.根據MOFs中金屬節點、有機配體和孔隙空間的3個基本元素,實現MOFs基M(bpy)/M(phen)催 化 劑 組 裝 的3種 方 法,如 圖1所 示:1)M(bpy)/M(phen)部 分 支 撐 在 金 屬 節 點 上;2)將M(bpy)/M(phen)部分結合到有機連接體上;3)將M(bpy)/M(phen)部分封裝在孔隙空間中.對于MOFs孔隙空間內的M(bpy)/M(phen)部分的封裝,可以使用3種成熟的方法,包括原位封裝、合成后引入和空腔內運輸[4].
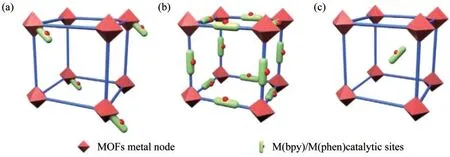
圖1 活性M(bpy)/M(phen)在MOFs框架中的位置.
以2,2’-聯吡啶-5,5’二羧酸(H2bpydc)配體為例,研究了不同的配體對H2bpydc的影響[5].圖2列舉了MOFs基M(bpy)催化劑的制備方法,包括直接摻入/封裝和組裝合成后修飾(PSM)策略.

圖2 MOFs基M(bpy)催化劑的合成方法示意圖(以H2bpydc配體為例).(a)直接封裝;(b)混合配體封裝;(c)原位封裝;(d)溶劑輔助配體交換;(e)離子交換;(f)配體在金屬簇上的修飾;(g)直接合成后金屬化;(h)組裝后修飾
2 M(bpy)/M(phen)的應用
M(bpy)/M(phen)部分可以通過直接或合成后修飾整合到MOFs框架中.通過直接摻入方法將M(bpy)/M(phen)部分固定到MOFs中主要限于惰性第二或第三排過渡金屬,而那些在溶劑熱條件下難以保持完整性的不穩定金屬化合物(尤其是第一排過渡金屬物種),應通過合成后修飾的方法修飾在MOFs中.其中,含有bpy配位配體的H2bpydc通常用于各種實例,M(bpy)已經成功修飾在MOFs中用于催化,其中M可以表示錳(Mn2+)、二價鐵離子(Fe2+)、鈷離子(Co2+)、鎳離子(Ni2+)、銅離子(Cu2+)、鋅離子(Zn2+)、釕離子(Ru2+)、銠離子(Rh2+)、鈀離子(Pd2+)、銀離子(Ag+)、錸離子(Re+)、鉑離子(Pt2+)、銥離子(Ir3+)和四價鈰離子(Ce4+).特別是,將M(bpy)/M(phen)部分作為催化位點加入到鋯(Zr(IV))基UiO系列MOFs中,由于其高度穩定的結晶性和優異的穩定性,已成功應用于許多催化反應[6-7].
2.1 Mn(bpy)用于二氧化碳(CO2)光還原催化劑
羰基錳UiO-67材料(Mn(bpy)(CO)3Br)絡合物是一種高效的CO2還原光催化劑.如圖3(a)~3(c)所示,Mn(bpydc)(CO)3Br已通過PSM方法固定在bpy修飾的MOF UiO-67中,有效避免了在均相催化系統中經常形成的非活性Mn-Mn二聚反應的產生[8].以[Ru(dmb)3]2+(dmb表示4,4’二甲基-2,2’-聯吡啶)和1-芐基-1,4-二氫煙酰胺為光敏劑和犧牲劑,制得催化劑UiO-67-Mn(bpy)(CO)3Br,其表現出優異的CO2光還原催化性能,轉換頻率(TOF)高達110次,甲酸選擇性96%,高于已報道的均相類似物和許多摻雜貴金屬的MOFs光催化劑.活性的增加歸因于UiO-67的框架結構,它可以抑制還原Mn(bpy)部分的二聚失活,從而提供分離的催化活性中心.此外,這種MOFs基催化劑的穩定性使其具有高度的可回收性和可重復使用性.
2.2 Fe(bpy)催化的醇氧化反應
2017年,XIN等[9]用六水合氯化鐵(FeCl3·6H2O)對bpy進行修飾,得到了MOF Fe@UiO-67-bpydc,制備出了Fe(bpy)功能化的MOF催化劑.在氧氣(O2,1 atm)存在下,可以用作促進醇氧化的有效催化劑.該多相催化體系可負載多種官能團,甚至雜環芳香醇.大體積醇的轉化率顯著降低,這是由于分子通過活性中心進入材料內部時被限制擴散所致.此外,該催化劑在好氧芐基氧化反應中也具有良好的適用性.熱過濾試驗和對溶液的電感耦合等離子體原子發射光譜儀(ICP-AES)分析發現,Fe浸出可以忽略不計,顯示出更好的穩定性.經過5次循環試驗后,催化性能基本保持不變,顯示出優異的可循環性和可重復使用性.
2.3 Co(bpy)/Co(phen)催化的炔烴加氫反應
ZHANG等[10]通過bpy和酚基Zr-MOF的PSM金屬化制備了一系列含Co(bpy)/Co(phen)的Zr-MOF催化劑,如圖3(d)~3(e)所示.其中,得到的催化前體Bpy-UiO-CoCl2在四氫呋喃(THF)中被三乙基硼氫化鈉(NaBEt3H)活化,得到活性Bpy-UiO-Co(THF)2.這種催化劑對于烯烴加氫反應,顯示出非常高的周轉數(TON))和周轉頻率(TOF).對催化機理的深入研究證明,活性物種(bpy·-)CoI(THF)2對烯烴加氫催化循環的啟動起著重要作用.這些活性催化劑還有效地催化了末端和內部烯烴的硼氫化反應.活性和穩定性的顯著增強源于固定在MOFs連接體上的高度分散的分離活性Co催化位點.此外,Bpy-UiO-Co(THF)2可以多次回收和再循環.
2.4 Ni(bpy)催化的乙烯二聚反應
MADRAHIMOV等[11]通過水熱法,將Ni(bpy)部分錨定在MOF-NU-1000的金屬節點上.與氯化鎳(NiCl2)反應后,可以觀察到明顯的顏色變化,從NU-1000-bpy的橙色變為綠色,生成NU-1000-bpy-NiCl2.經氯化二乙基鋁(Et2AlCl)預處理后,其在室溫下對乙烯二聚反應表現出優異的催化性能和可重復使用性,優于相應的均相對應物聯吡啶氯化鎳(Ni(bpy)Cl2),表明分子催化劑在MOFs中錨定后的催化性能具有巨大潛力.值得注意的是,NU-1000中的中孔(3.1 nm)和微孔(1.6 nm)通道有利于連續流動條件下的氣相反應.通過疊氮化物-炔環加成反應,MADRAHIMOV等[11]成功地將Ni(bpy)部分固定在疊氮化物官能化的MOF UiO-66-N3上,形成UiO-66-Ni(bpy),如圖3(f)所示.在Suzuki-Miyaura反應中,含量僅為3%(摩爾分數)Ni的非均相Ni(bpy)基催化劑就使反應的產率達到了96%,而相應的均相催化劑的產率僅為7%.此外,在至少7個連續循環中,催化劑的穩定產率為96%,顯示出良好的可回收性和可重復使用性.

圖3 Mn@UiO-67,Co@UiO-67,Ni@UiO-67材料的合成與應用.(a)Mn(bpy)的合成;(b)光催化實驗期間甲酸鹽周轉數的曲線圖;(c)催化甲酸鹽形成的機理探究;(d)具有Co(bpy)/Co(phen)部分的Zr基MOF催化劑的示意圖;(e)MOFs中活性位點示意圖;(f)UiO-66-N3的結構和其催化的Suzuki-Miyaura反應
2.5 Cu(bpy)催化的環辛烯的選擇性氧化反應
2015年,TOYAO等[12]通過PSM將溴化銅(CuBr2)錨定在bpy改性的UiO-67的bpy單元上,以開發高效且可回收的催化劑.得到Zr-MOF-bpy-CuBr2催化劑,這種催化劑可以在450℃以下保持結構完整性.材料的孔徑為(7?),環辛烯分子(5.5?)可以通過通道擴散,因此可以在通道中獲得更多的活性位點.在Cu負載量為3.3%的條件下,環辛烯的選擇氧化反應得到了比相應的均相產物Cu(bpy)Br2高84.3%的產率.非均相材料的催化位點可以有效防止催化劑失活以及金屬二聚體的產生,從而提高催化穩定性.連續運行3次后,材料結構保持完整.
2.6 Zn(bpy)催化的芳炔基苯胺的分子內氫胺化反應
類似地,LI等[13]在bpy功能化UiO-67的bpy單元上引入了四氟硼酸鋅(Zn(BF4)2),通過PSM策略構建了一種高效、穩定的單中心催化劑Zn-UiO-67-bpy,如圖4所示.在沒有其他配體(堿或酸)的情況下,Zn-UiO-67-bpy可以催化芳炔基苯胺的分子內氫胺化反應,以優異的產率獲得目標產物吲哚,而在相應的均相催化體系中得到了不需要的1-(2-氨基苯基)-2-苯乙酮,產率為73%~86%.與均相催化體系相比,MOFs基催化劑提供了一個特殊的內部局部環境,從而產生了不同的主要產物.此外,MOFs基催化劑具有較大的底物范圍,可循環使用至少5次,而不會顯著減少活性和結構破壞.值得注意的是,大多數用于鄰炔基苯胺雜環化的非均相金屬基催化劑涉及貴金屬(如金(Au),Ag和Pt).因此,貴金屬催化的反應也可以通過使用MOFs負載的堿金屬催化劑來實現.

圖4 (a)Zn-UiO-67-bpy的合成和(b)鄰炔基苯胺的催化分子內氫胺化反應
2.7 Ru(bpy)催化的好氧及光氧化反應
2011年,WANG等[14]通過一鍋法,成功地將Ir(bpy),Re(bpy)和Ru(bpy)部分錨定到MOF-UiO-67中,如圖5所示.所有改性的MOFs均具有較高的比表面積,熱穩定性高達400℃.Ru(bpy)3摻雜MOFs催化劑在3種有機轉化(Aza-Henry反應、好氧胺偶聯和硫苯甲醚的好氧氧化)中表現出優異的光催化性能,產率分別達到97%,91%和73%.此外,在重復使用性試驗中,MOFs催化劑可循環使用至少3次,且無明顯的活性損失及Ru2+浸出和骨架損壞,這證實了MOFs基催化劑具有優異的穩定性.
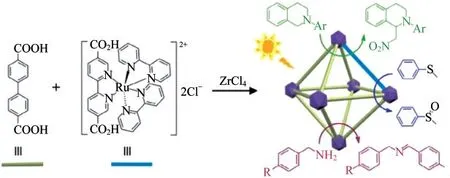
圖5 Ru(bpy)3摻雜的UiO-67催化的Aza-Henry反應、好氧胺偶聯反應和硫苯甲醚的光氧化
2.8 Rh(bpy)電催化CO2還原反應
Cp*Rh(bpy)Cl2(Cp*是五甲基環戊二烯)已被證明可通過電催化將CO2還原為甲酸鹽.考慮到衍生的Cp*Rh(bpydc)Cl2可以作為有機配體,并且多金屬氧酸鹽(POM)具有顯著的氧化還原和酸堿性質.BENSEGHIR等[15]首次將Rh絡合物和Keggin型磷鎢酸鹽(PW12O403-(PW12)POMs)固定到Zr(IV)基MOFUiO-67(PW12,Cp*Rh)@UiO-67中,如圖6(a)所示.Cp*Rh為活性位點,分別以[Rh(bpy)3]Cl2和三乙醇胺(TEOA)為光敏劑和犧牲劑,與Cp*Rh@UiO-67進行比較.發現甲酸鹽和氫氣(H2)產量分別提高了2倍和2.5倍,這比相應的均相催化劑產率高了很多,如圖6(b)和6(c).
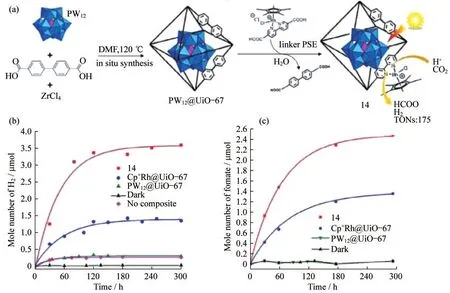
圖6 以磷鎢酸鹽為內核的Cp*Rh@UiO-67材料的制備與應用.
2.9 Ag(bpy)催化的末端炔烴羧化反應
由于特殊的d10電子構型,Ag+能有效地活化碳碳三鍵,形成具有活性的活性Ag-π鍵化合物.2019年,GONG等[16]通過PSM方法將Ag(bpy)固定到UiO-66@UiO-67的核-殼異質結構,如圖7所示.核-殼結構提供了活性Ag(bpy)的良好分散性以及基質和Ag(bpy)之間的有效相互作用.獲得的UiO66@UiO-67-BPY-Ag是CO2與末端炔烴羧化反應的高效催化劑.在溫和的條件下,核殼結構MOFs催化劑比單一MOFs催化劑能獲得更高的產率,這歸因于核-殼可以實現協同效應.此外,該材料可循環使用5次,損失和金屬浸出可忽略不計.

圖7 核殼MOFs材料的制備及CO2與末端炔烴的羧化反應示意圖
總之,與均相分子催化劑相比,固定在MOFs框架內的活性M(bpy)/M(phen)絡合物不僅能有效抑制非活性低聚物的形成,而且提供了一種新的快速能量和電子轉移路徑,從而提高了催化性能和選擇性.相比均相催化劑來講,文章提到的Fe(bpy)催化的醇氧化反應、Cu(bpy)催化的環辛烯的選擇性氧化反應等都擴大了MOFs材料的優越性,在一定程度上大幅提升了反應的產率及催化活性;在電化學中,Rh(bpy)催化的CO2還原反應比傳統的催化劑性能提升了2倍以上;在光反應中,以Ru(bpy)和Mn(bpy)為催化劑的體系中反應結果都相比傳統催化劑有優勢.
3 總結與展望
綜上所述,催化活性M(bpy)/M(phen)部分可以固定在金屬節點和配體上,或者通過直接和合成后的修飾將其封裝到MOFs結構中.bpy/phen修飾的MOFs依靠活性金屬離子的種類和MOFs的載體,在多相催化領域具有普適性.高度分散的孤立金屬位點,以及MOFs的固有特征,包括高比表面積和永久孔隙率,在有機轉化等非均相體系中表現出優異的催化活性.雖然在M(bpy)/M(phen)功能化MOFs方面取得了一些顯著的進展和成果,但MOFs基金屬催化劑的研究仍處于起步階段,還有一些問題需要解決.例如:溶劑熱合成條件苛刻、反應時間長、溫度高、壓力大,難以大規模生產;大多數MOF穩定性較差,極大地限制了其在催化領域的實際應用.目前,只有一些經典的、易于合成且相對穩定的MOFs被廣泛用于固定活性M(bpy)/M(phen)部分,如Zr基UiO系列和Al基MOF-253 MOF.此外,在材料的制備過程中應考慮先進的表征技術,尤其是原位表征技術,包括原位紅外光譜(IR)、原位X射線光電子能譜(XPS)和原位X射線吸收精細結構譜(EXAFS),以監測反應過程,從而識別反應中間體,探索活性中心并揭示結構-活性關系,這將為進一步設計高效的M(bpy)/M(phen)功能化MOFs/COFs催化劑提供深入的見解.
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
