石墨烯基氮摻雜層級孔碳負載鈀及其PTA加氫精制應用
2022-11-18 04:05:26張曉利楊葉偉何觀偉
工業催化 2022年10期
殷 迪,鄭 晴,張曉利,卞 雯,楊葉偉,何觀偉
(西北化工研究院有限公司,陜西 西安 710061)
精對苯二甲酸(PTA)是一種重要的有機二元酸,廣泛用于聚酯纖維、聚酯瓶片和聚酯薄膜的生產。PTA工業化生產的典型工藝為兩步法精PTA工藝,首先以對二甲苯(PX)為原料,在催化劑作用下氧化制得粗對苯二甲酸。粗對苯二甲酸中含有的副產物4-羧基苯甲醛(4-CBA),會影響PTA產品質量以及后續聚酯的加工性能,需要采用加氫精制步驟除去。PTA的加氫精制反應是指在鈀炭催化劑作用下,在溫度(270~290) ℃、壓力(7.0~8.0) MPa條件下,將粗對苯二甲酸中的4-CBA還原成易溶于水的對甲基苯甲酸,再經結晶、分離、干燥制得纖維級PTA[1-3]。
鈀是粗對苯二甲酸加氫精制使用最廣泛的活性組分,工業化生產中常用的催化劑為活性炭負載的鈀催化劑,但仍存在一些挑戰。一方面,活性炭因其中巨大的微孔結構不利于分子的擴散,必須精心設計碳材料的多孔結構以強化傳質從而提高催化效率;另一方面,由于活性炭其表面化學惰性,與金屬的相互作用較弱,活性金屬物種在活性炭上表現出較大的聚集和浸出傾向,導致活性金屬粒子的流失和團聚。因此,研究者嘗試對載體進行修飾,以增強這種相互作用。
層級孔碳材料是一類新型的多孔碳材料[4-8],同時兼具不同尺寸與功能的微孔、中孔或大孔。層級孔碳材料是較理想的金屬催化劑載體,其大孔可以阻礙所負載顆粒的團聚,介孔提供了反應溶液的輸運通道,微孔促進了生成氣體的擴散,大比表面積有助于催化活性位和反應物的接觸。一般來說,純碳材料的親水性較差,而這在一定程度上掣肘了其在各個方面的應用。在元素周期表中氮原子位于碳原子相鄰位置,因而可取代碳材料骨架中的碳原子,摻雜在碳材料中的氮原子可以在材料表面結構的改變、孔道結構的調節、親水性的增強、對材料表面pKa值的影響、材料電子傳輸速率的改善等方面起到極大作用,從而拓展碳材料的應用范圍[9-11]。碳材料中摻雜的N原子可有效地提高催化劑的催化活性以及催化劑的壽命,顯著改善其吸附及催化性能;有利于降低貴金屬的用量,減少催化劑制備成本;提高碳材料對氫氣的吸附能力。
本文以氧化石墨烯為結構導向劑,間苯二酚-甲醛-三聚氰胺為前驅體,其中三聚氰胺為氮源,經溶膠-凝膠法及炭化制備得到三維堆疊石墨烯基氮摻雜層級孔碳納米片(NCNS),負載Pd后制得的Pd/NCNS催化劑用于PTA加氫精制,探索NCNS孔道結構及氮含量的調控機制,重點考察負載Pd分散度與氮含量的相關性以及載體孔道結構及氮含量對催化活性的影響。
1 實驗部分
1.1 原料
氧化石墨烯(GO),12 mg·mL-1,中國科學院山西煤炭化學研究所;甲醛(37%)、苯二酚、三聚氰胺、氯鈀酸鈉、鹽酸(37%)、氫氧化鈉、甲酸鈉,分析純;Pd/C催化劑,0.5%;粗對苯二甲酸,含4-CBA質量分數3%,中國石化上海石油化工研究院。
1.2 催化劑制備
1.2.1 碳納米片制備
將一定質量的間苯二酚(R)與甲醛溶液(F,質量分數37%)混合(R與 F的物質的量比為1∶2),在40 ℃的恒溫水浴中反應30 min,得到RF溶液。將三聚氰胺(M)在80 ℃溶解于甲醛溶液(F)中攪拌15 min(M與F的物質的量比為 1∶3),得到MF溶液。將RF和MF溶液混合,即得到一定濃度的RMF溶液,加入一定量經過超聲處理的氧化石墨烯水分散液(RMF與GO 的質量比分別為 40、60、80、100),將所得的混合液在80 ℃水浴中攪拌24 h,抽濾、80 ℃干燥,得到聚合物RMF-GO粉末。
將所得的聚合物氧化石墨烯納米片在N2氣流保護下,以1 ℃·min-1的升溫速率升至800 ℃,并停留3 h,即可得到含氮多孔炭-氧化石墨烯納米片材料,命名為NCNS-x(y),其中x為M與R物質的量比,y為RMF和氧化石墨烯質量比。
1.2.2 Pd的負載
稱取含氯化鈀4.2 g的氯鈀酸鈉溶液23.1 g,加入200 g去離子水配制成浸漬膠液,調節浸漬膠液pH=4.2,5 min內將溶液倒在500 g碳載體上,并于室溫靜置12 h。浸漬后的催化劑前驅體在空氣中老化6 h。取200 g濃度為1%的甲酸鈉溶液配制成水溶液,水溶液的量以剛好浸沒所有催化劑前驅體為宜,在120 ℃還原120 min,將負載于碳載體上的Pd化合物還原成金屬Pd,然后用去離子水洗滌,至溶液呈中性或無氯離子為止。過濾后即得Pd負載質量分數為0.5%的Pd/NCNS催化劑。
1.3 催化劑表征
孔道結構在美國康塔公司(Quadrasorb SI)全自動氮氣吸附儀上于77 K條件下進行測定,采用BET方法計算樣品的比表面積,QS-DFT方法計算孔徑分布,在吸附量最大值處計算總孔容,采用T-plot法計算微孔表面積及微孔孔容。
采用日本電子株式會社7100F型場發射掃描電子顯微鏡(SEM)觀察樣品的表面形貌。
樣品中的C、N、H元素含量由德國Elementar Vario ELⅢ 元素分析儀測得,O元素的含量則采用差量法計算。
X射線衍射圖譜由日本理學公司 D/max 2550型X射線衍射儀測定,CuKα,λ=0.154 06 nm,工作電流為40 mA,工作電壓為40 kV,以0.010°·s-1速率進行掃描。
樣品的表面組成采用瑞典VG Scientific公司的ESCALab220i-XL型光電子能譜儀測定,激發源為單色化A1K X射線,功率為300 W,分析時的基礎真空為3×10-7Pa。
采用H2-O2滴定法測定樣品的Pd分散度、活性表面積和晶粒度,在美國麥克儀器公司AutoChemⅡ2920化學吸附儀上進行。樣品首先在氫氣氣氛中200 ℃還原處理,再切換惰氣氣氛吹掃至基線平穩,切換氧氣進行氧化,然后用脈沖氫氣還原氧化物,根據耗氫量即可計算表面金屬鈀的原子數,進一步計算得到Pd的分散度、活性表面積和晶粒度。
1.4 催化劑活性評價
催化劑活性評價在不銹鋼攪拌間歇高壓反應釜中進行,評價條件為:催化劑裝填量為2.0 g,粗對苯二甲酸30.0 g,含4-CBA為1.0 g,水溶液900.0 mL,反應壓力7.5 MPa,反應溫度280 ℃。反應后液體產物通過高效液相色譜配紫外檢測器進行定量分析,通過計算剩余的4-CBA含量評價催化劑活性,剩余的4-CBA含量越低,說明催化劑的催化加氫效率越高。
2 結果與討論
2.1 碳載體制備
采用溶膠-凝膠法結合結構導向支撐骨架制備氮摻雜碳納米片,關鍵點在于如何在二維結構導向劑氧化石墨烯(GO)表面均勻地包覆樹脂聚合物,從而形成典型的三明治多層結構。因為氧化石墨烯表面具有羥基等大量的親水性官能團,GO可以穩定地分散于水中,形成均勻的水分散液。而間苯二酚-甲醛-三聚氰胺(RMF)水溶液其表面則呈現出一定的正電荷性質,當二者混合時,可以通過靜電引力作用而使得RMF酚醛樹脂在GO表面進行快速聚合反應,進而形成層狀包覆結構。通過調節GO和RMF酚醛樹脂的質量比,即可制得具有不同包覆厚度的納米片層結構。而通過調節前驅體中三聚氰胺和間苯二酚物質的量比,可以控制碳納米片的氮含量。
2.1.1 RMF與GO質量比
通過改變結構導向劑GO和包覆聚合物的質量比,制備得到具有不同包覆厚度的納米片層結構。圖1為不同RMF與GO質量比條件下得到的樣品炭化后的掃描電鏡照片。

圖1 不同RMF與GO質量比的氮摻雜碳納米片的掃描電鏡照片Figure 1 SEM images of nitrogen doped carbon nanosheets with various mass ratio of RMF/GO
由圖1可以明顯看出,隨著RMF與GO質量比增大,納米片層結構的厚度依次增加,當RMF與GO質量比為40時,片層厚度約為20 nm;當RMF與GO質量比為60時,片層厚度約增加到50 nm;而當RMF與GO質量比提高到80和100時,片層厚度約為60 nm和90 nm。因而,通過改變聚合物添加量,可以有效地對納米片層厚度進行調節。值得注意的是,整個體系中RMF樹脂在氧化石墨烯表面包覆均勻,沒有自我聚合而形成聚合物微球,表明GO對其起到了優異的結構導向作用。
圖2為不同RMF與GO質量比得到的碳納米片的氮氣吸附-脫附等溫線和DFT孔徑分布。從圖2可以看到,所有的樣品在較低的相對壓力下,均具有較大的氮氣吸附量,表明材料中存在一定數量的微孔,同時隨著聚合物用量的增大,微孔比例也逐漸增大(表1)。但當聚合物用量較低時,脫附曲線具有典型的H2型滯后環,表現出一定程度的類石墨烯的結構,即狹窄的片層堆積而成的孔道結構。隨著RMF聚合物用量增加,納米片層厚度不斷增大,更明顯地表現為純微孔結構,其H2滯回環也逐漸消失。表明納米片層最終都表現為RMF聚合物的分子鏈接形成的孔道,氧化石墨烯對孔結構的貢獻則越來越小。
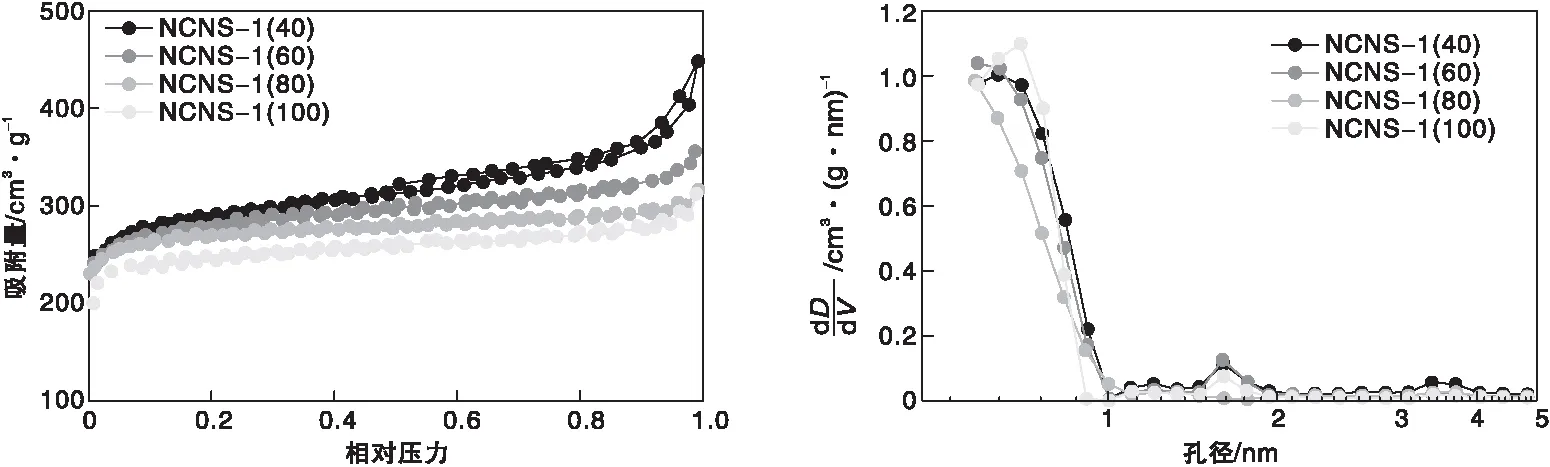
圖2 不同RMF與GO質量比氮摻雜碳納米片的氮氣吸附-脫附等溫線和孔徑分布Figure 2 N2 adsorption-desorption isotherms,and DFT pore size distribution of nitrogen doped carbon nanosheets with various mass ratio of RMF/GO
樣品孔結構參數如表1所示。由表1可以看出,隨著RMF與GO質量比增大,樣品的比表面積均在約1 000 m2·g-1波動,微孔比表面積則在約950 m2·g-1波動,兩者的變化均不明顯。采用差減法可知中孔孔容則從0.32 cm3·g-1逐漸減小到0.16 cm3·g-1、0.12 cm3·g-1和0.11 cm3·g-1,正好對應圖2脫附曲線H2滯回環面積的逐漸減小。
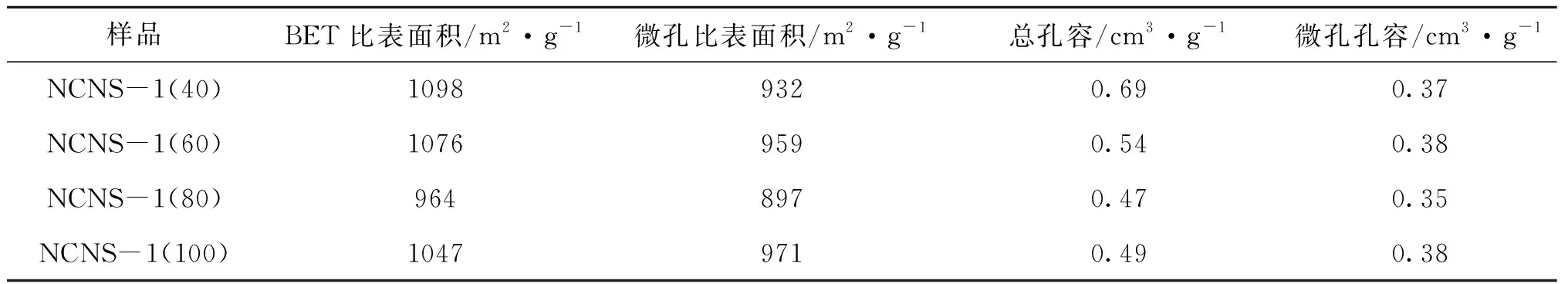
表1 不同RMF與GO質量比氮摻雜碳納米片的孔結構參數
表2是不同RMF與GO質量比氮摻雜碳納米片的CNH元素分析結果。從表2可見,隨RMF與GO質量比增加,氮含量從NCNS-1(40)的9.14%緩慢增加到NCNS-1(100)的10.52%,碳元素的含量呈現緩慢增加趨勢,氫元素基本持平,而氧元素則從NCNS-1(40)的8.56%逐漸減小到NCNS-1(100)的4.73%。這是因為前驅體RMF-GO中GO的含氧量比RMF高,因此,前驅體中GO比例越小,碳化后碳納米片的含氧量也越小;反之,氮元素幾乎全部來自RMF中的三聚氰胺,隨著前驅體中RMF比例增大,碳化后碳納米片的氮含量也逐漸增大,碳元素含量隨之提高。但由于RMF中M與R的比例維持在1∶1,因此樣品的氮碳比接近0.13。

表2 不同RMF與GO質量比氮摻雜碳納米片的元素組成
2.1.2 三聚氰胺含量
圖3為不同M與R物質的量比的氮摻雜碳納米片的掃描電鏡照片。從圖3可以看出,三個樣品均呈現相類似的片層結構,片層厚度約20 nm,表明M與R物質的量比對氮摻雜碳納米片的形貌沒有明顯影響。

圖3 不同M與R物質的量比氮摻雜碳納米片的掃描電鏡照片Figure 3 SEM images of nitrogen doped carbon nanosheets with various mole ratio of M/R
圖4為不同M與R物質的量比得到的氮摻雜碳納米片的氮氣吸附-脫附等溫線和DFT孔徑分布。由圖4可見,所有樣品的氮氣吸附-脫附曲線在相對壓力較低時呈現快速上升趨勢,接著吸附曲線變得相對平坦,表現出Ⅰ型吸附等溫線特征(按IUPAC分類),說明樣品呈現出典型的微孔結構。同時可以觀察到,在M與R物質的量比為0.5、1和2時,樣品在低相對壓力處的吸附量類似,這反映在樣品的微孔比表面積從M與R物質的量比為0.5時的865 m2·g-1增加到M與R物質的量比為1時的932 m2·g-1和M與R物質的量比為2時的1 003 m2·g-1(表3),因前驅體中氮含量愈高,聚合物分解速率越快,反應越劇烈,隨著分解程度的提高產生出更多微孔。M與R物質的量比為0.5和1的樣品存在明顯的H2型滯回環,M與R物質的量比為0.5的面積更大,說明在這兩個樣品中存在典型的類石墨烯結構,而M與R物質的量比為2的樣品則無明顯的H2滯回環,呈現單純微孔結構,采用差減法得三個樣品中孔孔容分別為0.72 cm3·g-1、0.32 cm3·g-1、0.07 cm3·g-1,呈逐漸下降趨勢,這是因為,隨著分解劇烈程度的提高,片層之間黏連堆疊的程度也相應提高,導致片層間中大孔減少。
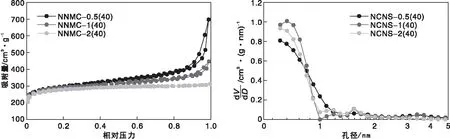
圖4 不同M與R物質的量比氮摻雜碳納米片的氮氣吸附-脫附等溫線和孔徑分布Figure 4 N2 adsorption-desorption isotherms and DFT pore size distribution of nitrogen doped carbon nanosheets with various mole ratio of M/R
表4為不同M與R物質的量比氮摻雜碳納米片的CNH元素分析結果。從表4可見,隨著M與R物質的量比增加,氮含量呈快速增加趨勢,M與R物質的量比為0.5、1、2時的氮含量分別為6.38%、9.14%和13.49%。碳含量則呈減少趨勢,從82.82%逐漸減少到73.19%。氮碳比從0.09增加到0.20,也增加兩倍。說明通過調控M與R物質的量比,可以實現摻氮量可控的碳納米片制備。
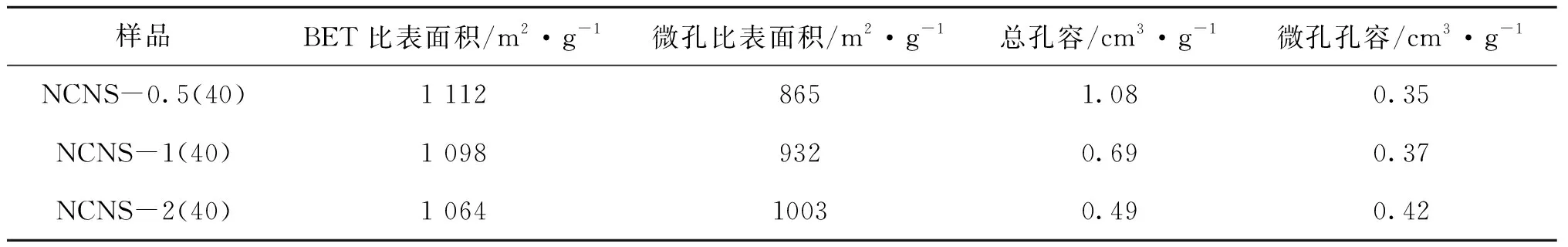
表3 不同M與R物質的量比氮摻雜碳納米片的孔結構參數
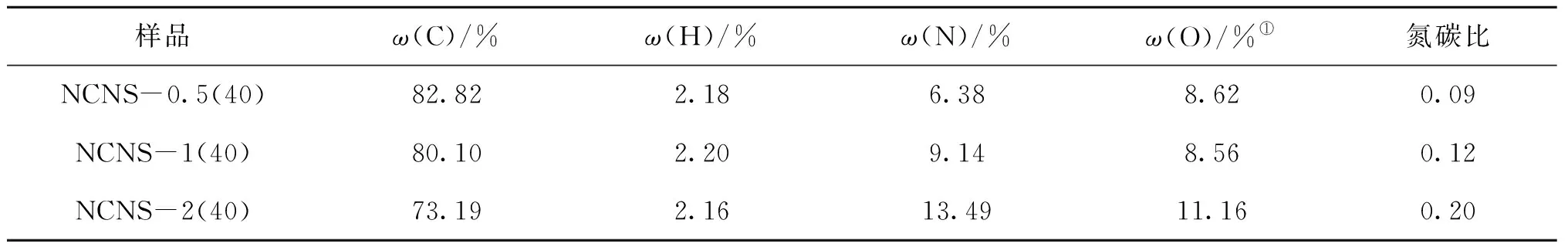
表4 不同M與R物質的量比氮摻雜碳納米片的元素組成
圖5為不同M與R物質的量比氮摻雜碳納米片的XPS分析結果。由圖5可知,XPS全譜掃描曲線中出現明顯的N1s信號峰,說明N元素已被成功摻雜到碳納米片的碳骨架結構中。與此同時,隨著M與R物質的量比增加,樣品的N1s信號峰強度逐漸增強,表明碳納米片中摻雜的N元素含量可以通過改變M與R物質的量比調控。由圖5還可以看出,所有氮摻雜碳納米片的N1s峰均可在結合能為398.6 eV、399.9 eV和400.9 eV處分為三個峰,分別對應于吡啶氮(N-6)、吡咯氮(N-5)和石墨化氮(N-Q)。吡啶氮(N-6)以六元環的結構形式存在于碳層石墨環的邊緣,并與兩個碳原子相連,在其外層電子軌道存在一對未成鍵孤對電子,因而呈現出強的堿性。吡咯氮(N-5)則以五元環結構形式存在于碳層石墨環的邊緣,其為石墨環的離域π系統供應了一對P電子,從而強化了碳材料對小分子氣體的界面吸附能力。石墨化氮(N-Q)位于石墨烯片層的中心位置,其以sp2雜化軌道結構形式與三個碳原子相連,同時參與了石墨六元環中離域大π鍵的形成,從而強化了石墨烯片層表面的堿性。
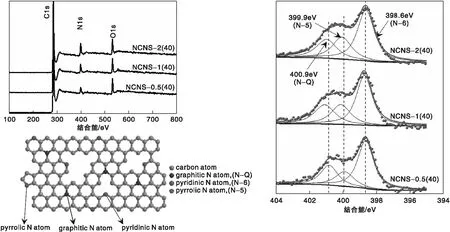
圖5 不同M與R物質的量比氮摻雜碳納米片的XPS全譜掃描曲線、XPS N1s及其分峰結果、不同氮種類示意圖Figure 5 XPS survey,XPS N1s and peak division,and schematic diagram for different nitrogen species of NCNSs with various mole ratio of M/R
2.2 Pd負載量
圖6給出氮摻雜碳納米片負載鈀的Pd/NCNS及商用Pd/C的XRD圖。
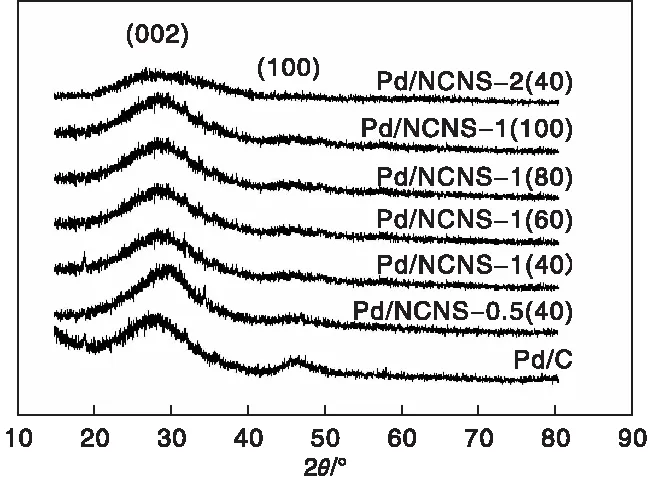
圖6 氮摻雜碳納米片負載鈀及商用Pd/C的XRD圖Figure 6 XRD patterns of Pd/NCNSs and commercial Pd/C
由圖6可以看出,所有樣品的出峰位置以及峰形基本保持一致,其在2θ=23.7°以及43.6°兩處均呈現出異常彌散的衍射峰,這兩個峰分別對應于石墨微晶結構的[002]和[100]晶面,表明樣品均呈現出典型的無定型炭結構。值得注意的是,隨著M與R物質的量比即氮含量的增加,樣品對應于石墨[002]晶面的峰形漸漸變寬,同時峰位置向左輕微逐漸偏移,說明樣品的石墨化度呈現降低的趨勢,而且類石墨微晶層間距逐漸輕微變寬。在XRD圖中沒有觀察到歸屬Pd晶體的衍射峰,說明此時Pd納米顆粒被高度分散在載體表面,且其晶粒尺寸非常小,超出XRD儀器的檢測限。
因為XRD圖譜沒有給出Pd晶粒的特征峰,為了表征Pd晶粒分散度、活性比表面積和晶粒度,采用H2-O2滴定法進行了上述工作,結果示于表5。由表5可知,氮摻雜碳納米片負載Pd的分散度要顯著高于商用Pd/C,具有較大活性比表面積和較小晶粒度;值得注意的是,隨著氮含量的提高,Pd分散度也明顯上升。這些結果說明氮摻雜有助于Pd在碳載體表面的分散。由表5還可以看出,負載Pd后,所有樣品的比表面積都有小幅度的降低,平均孔徑為(0.61~0.67) nm,這是N2吸附的檢測下限,而商用Pd/C的平均孔徑卻為2.5 nm,這是因為碳納米片只經過碳化,而商用Pd/C的活性炭載體經過了活化步驟。由于選擇DFT計算方法,碳納米片堆疊形成的介孔及大孔無法在此處體現。
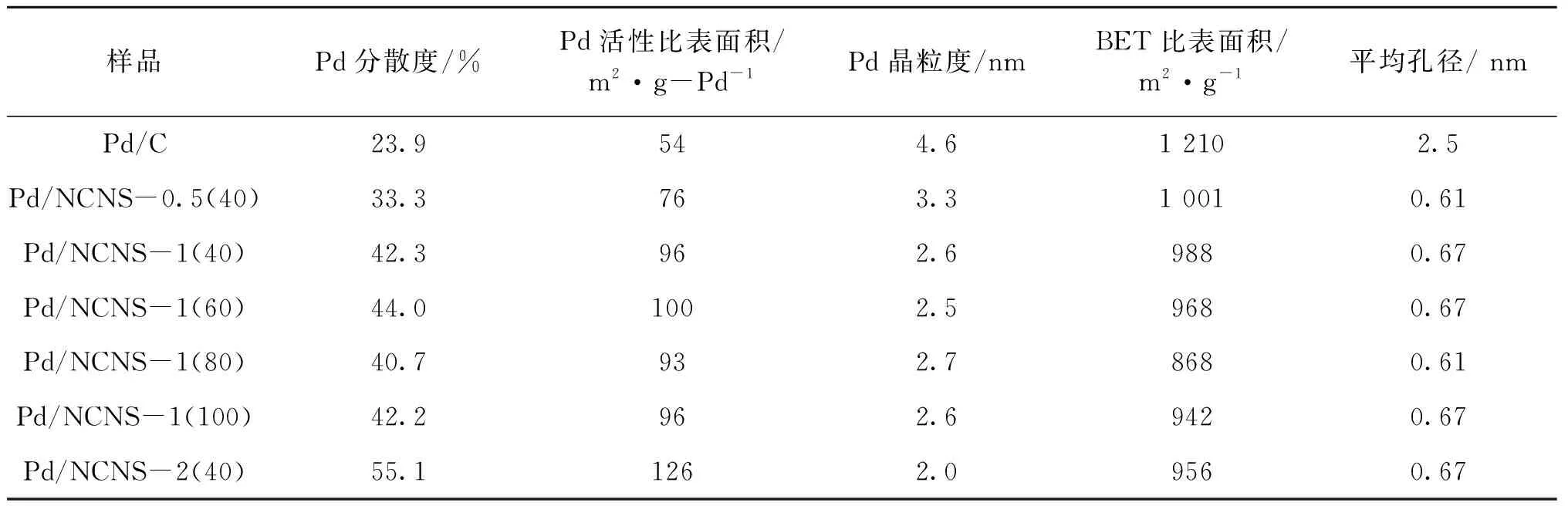
表5 氮摻雜碳納米片負載鈀及商用Pd/C的H2-O2滴定結果及孔結構參數
2.3 活性評價結果
樣品的活性評價數據及相關聯的表征參數列于表6。由表6可知,4-CBA轉化率隨樣品氮含量及Pd分散度的增加而提高。商用Pd/C催化劑不含氮,Pd分散度僅為23.9%,4-CBA轉化率為96.3%;而氮含量最高的Pd/NCNS-2(40)催化劑,其Pd分散度也是樣品中最高的(55.1%),4-CBA轉化率達99.6%。這是因為氮摻雜所致的Pd分散度愈高,暴露在表面的活性位愈多,同時氮還可能起到了吸附活化氫氣和4-CBA的作用。此外,對于含氮量類似,但片層厚度不同的Pd/NCNS-1系列樣品,隨著片層厚度增加,其活性呈緩慢下降趨勢,這可能是擴散的影響。氮摻雜碳納米片負載鈀催化劑活性高于商用Pd/C催化劑,其擴散通道較短也是重要的影響因素,活性炭的孔道由錯綜復雜的微孔構成,其長度遠大于碳納米片的厚度,而碳納米片堆疊而成的三維結構,片層間的中大孔起到了主要的傳質作用。
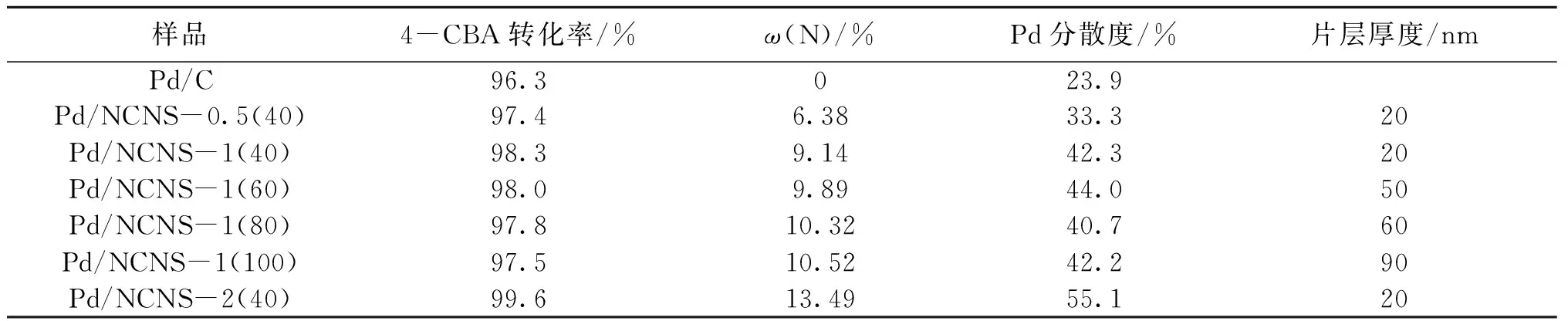
表6 氮摻雜碳納米片負載鈀催化劑及商用Pd/C催化劑的活性及相關表征參數
3 結 論
(1) 通過調節原料中間苯二酚-甲醛-三聚氰胺與氧化石墨烯的比例,經過溶膠-凝膠法及炭化可以制備出厚度均一可控的氮摻雜碳納米片材料,當RMF與GO的質量比分別為40、60、80、100時,碳化后樣品的片層厚度分別為20 nm、50 nm、60 nm、90 nm。通過控制三聚氰胺與間苯二酚物質的量比,可以調節碳納米片的氮含量。當M與R物質的量比分別為0.5、1和2時,所得碳納米片的氮含量分別為6.38%、9.14%、13.49%。
(2) 氮摻雜可提高碳載體上Pd分散度,不含氮的商用Pd/C催化劑上Pd分散度僅為23.9%,氮含量最高的Pd/NCNS-2(40)催化劑上Pd分散度可達55.1%。
(3) 氮摻雜碳納米片負載鈀催化劑表現出比商用Pd/C催化劑更高的活性,歸因于其較短的擴散通道、氮摻雜及其所致的高Pd分散度。
猜你喜歡
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
現代企業(2015年9期)2015-02-28 18:56:50
