T型鈣通道在心血管疾病中的研究進展*
2022-12-03 08:46:28張弛趙爽陳露嵐胡亦清申程
中國病理生理雜志 2022年11期
關鍵詞:小鼠
張弛,趙爽,陳露嵐,胡亦清,申程
(1濟寧醫學院附屬醫院,濟寧醫學院,山東 濟寧 272067;2上海市徐匯區中心醫院體檢科,上海 200031;3復旦大學附屬中山醫院心內科,上海市心血管病研究所,上海 200032;4濟寧醫學院附屬醫院心內科,濟寧市心血管疾病診療重點實驗室,山東 濟寧 272067)
電壓門控鈣通道(voltage-gated calcium channels,VGCC)能夠參與調節多種可興奮性細胞和非興奮性細胞的生理活動,其中T型鈣通道在神經元中產生低閾值鈣尖峰并影響動作電位放電模式,在神經遞質釋放、樹突共振現象及基因表達調控中起著關鍵作用[1]。在血管平滑肌細胞中調節肌源性張力,在內分泌細胞中調節激素的分泌,以及在精子中調節頂體反應。重要的是,T型鈣通道在心臟細胞中能夠影響心臟的起搏和脈沖的傳導[2-3]。近年來,對T型鈣通道活性的分子機制和作用靶點的研究不斷取得新的進展,轉基因動物實驗已經證明T型鈣通道是治療包括心肌肥大等多種心血管疾病的重要藥物靶點,其活性受多種激素的影響。因此,關注T型鈣通道在心血管疾病中的作用機制對于心血管疾病的治療和相關藥物的研發具有重要意義。
1 VGCC
VGCC激活并介導細胞外Ca2+內流進入細胞質。作為第二信使,Ca2+與許多細胞反應有關,包括神經遞質釋放、肌肉收縮和基因表達。因此VGCC是電興奮的關鍵信號傳感器,能將細胞膜上的電信號轉化為細胞內具有重要生理意義的Ca2+瞬態[4]。
1.1 功能心肌細胞膜上存在2種不同的VGCC(L型和T型)。VGCC具有3種依賴電壓的不同功能狀態:關閉或休眠通道、激活或打開通道和失活通道。去極化時,激活通道在幾毫秒內從靜止狀態轉變為激活狀態,然后迅速失活。然而,不同于Na+和K+主要通過膜電位改變的細胞膜流動交換方式,Ca2+通過VGCC進入細胞并與許多細胞反應相耦合。在心肌和平滑肌細胞中,VGCC通過增加細胞質中Ca2+濃度直接引發收縮而激活,或者通過肌漿網中對雷諾丁受體(ryanodine receptor,RyR)敏感的Ca2+釋放通道間接激活鈣依賴鈣釋放機制[5-6]。
1.2 亞型編碼VGCC的α1亞基目前已分出10種。按照α1亞基的基因型,可分為Cav1、Cav2和Cav3這3個家族,其中Cav1包含Cav1.1~1.4四個亞型,Cav2包 含Cav2.1~2.3三 個 亞 型,Cav3包 含Cav3.1~3.3三個亞型[7]。Cav1亞家族啟動神經元的收縮、分泌、調控基因表達,突觸的轉運整合和特殊感覺細胞帶狀突觸的傳遞。同時,Cav1在啟動心臟和平滑肌收縮中發揮重要作用,Cav1中含有一個形成孔的α1D亞基,是正常心臟起搏所必需的[8]。Cav2亞家族主要負責在快突觸中啟動突觸傳遞;Cav3亞家族則對于節律放電細胞(如心肌細胞和丘腦神經元)動作電位的重復放電非常重要[9]。
1.3 分類VGCC按照電壓激活特性分類,可分為高電壓激活型(high-voltage activation,HVA)和低電壓激活型(low-voltage activation,LVA),HVA通道在膜去極化程度大時響應激活,而LVA通道在低電壓變化時即可激活,此時的膜電位與可興奮性細胞靜息膜電位相近[10]。按照Ca2+電流門控特性分類,又可分 為L型(Cav1.1~1.4)、P/Q型(Cav2.1)、N型(Cav2.2)、R型(Cav2.3)和T型(Cav3.1~3.3)[10]。
2 T型鈣通道
2.1 結構和分子特性L型鈣通道是由α1核心亞基和附屬亞基(包括α2δ、細胞內β和跨膜γ)組成的異源四聚體。α1亞基由4組同源重復組成,每組重復包含6個跨膜片段(S1、S2、S3、S4、S5和S6)以及S5與S6之間形成的連接環。S5、S6及連接環共同形成通道的孔道,是藥物調節鈣通道的結合位點。每組重復中S4作為電壓感受域,在膜去極化時響應使通道打開[7],輔助亞基調節α1亞基的通道動力學、門控特性以及通道上膜運輸。2019年,顏寧團隊發表在《Nature》上的文章首次揭示了人類T型Cav3.1通道高分辨冷凍電鏡結構,提出與L型亞基不同的是,T型鈣通道的α1亞基可以自主支持通道功能。此外,由于Cav3.1的α1亞基上2個門控相關殘基不同于L型Cav1.1通道,只需更少的能量來激活通道胞內門控開放,促進Cav3.1在低電壓下激活。通道的高分辨率結構分析有助于對其獨特功能的機制研究,并通過引導通道激動劑或拮抗劑的結構研發藥物[11]。
Cav3亞家族傳導T型Ca2+電流。這種Ca2+電流在相對負的膜電位下被激活,與大多數細胞中的Na+電流范圍相同。而且與其他Ca2+電流相比,它們具有快速的電壓依賴性失活[12]。因為它們在負膜電位下被激活,而在負膜電位下Ca2+進入的驅動力很大。因此,這種Ca2+電流非常適合按一定的頻率觸發動作電位,它們同樣也很適合產生大量的Ca2+瞬態。通過cDNA克隆和測序鑒定得出Cav3是一個由3種α1亞基組成的家族。值得注意的是,這些鈣通道亞基具有與Cav1和Cav2通道相同的分子組織,但是只有25%的氨基酸序列相同,這表明鈣通道的亞家族在進化時就會彼此分離[9]。
盡管Cav3通道與Cav1和Cav2通道具有相似的結構,但目前沒有明確的證據表明它們與同一組的輔助亞基相互作用。事實上,普遍的觀點認為α1亞基獨立于其他亞基發揮作用,這在鈉通道和鈣通道家族中是獨一無二的[9]。
2.2 組織空間表達T型鈣通道開放時間較短,與低膜電位下Ca2+的跨膜運動有關,在竇房結(sinoatrial node,SAN)細胞早期除極(起搏電流)中起特殊作用。在胚胎期時,T型鈣通道在整個心臟廣泛表達,表明其在胎兒生命的特定階段的重要作用,但是出生后在心室肌細胞表達迅速下降。在成人心臟中,僅在心臟傳導系統中的起搏細胞可以觀察到高密度的T型鈣通道[13]。成人心臟SAN、房室結(atrioventricular node,AVN)和浦肯野纖維中保留Cav3.1和Cav3.2表達,提示其參與心臟自動性和傳導[14],并在SAN細胞早期除極中起到起搏電流的作用。研究表明,Cav3.2缺陷小鼠竇房節律正常,而Cav3.1缺陷小鼠SAN恢復時間延長,SAN細胞起搏器活動減慢,心率減慢,房室傳導延遲[15]。這些結果表明,Cav3.1是心臟心律產生的主要T型鈣通道參與者[16]。此外,在血管平滑肌中分布的T型鈣通道有維持血管平滑肌張力的作用[17]。
3 T型鈣通道與心血管疾病
3.1 T型鈣通道對心臟和血管功能的調節
3.1.1 T型鈣通道參與維持心臟自律性心臟自律性是指由于相對緩慢的舒張期去極化的存在,起搏細胞在SAN中產生自動化,它驅動膜電壓從復極化階段的結束直到到達下一個動作電位的閾值。之后SAN脈沖傳導到心房和AVN,并通過浦肯野纖維網傳播到心室,推動整個工作心肌的收縮[18]。心臟的起搏細胞主要位于SAN,將SAN起搏器細胞表面膜上離子通道的集合設想為一個表面的“膜時鐘”[19]。在舒張后期去極化過程中,肌漿網通過RyR產生釋放Ca2+,這些Ca2+被稱作是細胞內的“Ca2+時鐘”。在自動激發的SAN細胞中,膜時鐘和Ca2+時鐘不是單獨工作,而是通過膜電壓、肌膜下Ca2+、蛋白激酶A(protein kinase A,PKA)和磷酸化的鈣/鈣調蛋白依賴性蛋白激酶II(calcium/calmodulin-dependent protein kinase II,CaMKII)等相互作用,兩個子系統時鐘相互約束,形成一個耦合的時鐘模型,驅動正常心臟起搏器細胞的自動性[20]。SAN起搏器細胞同時表達T型和L型鈣通道。T型鈣通道的異構體Cav3.1和Cav3.2的mRNA在SAN中表達,成人SAN中主要的T型鈣通道亞型是Cav3.1。Cav3.1介導的鈣通道電流的激活閾值為-55 mV。然而,只有10%的穩態Cav3.1介導的鈣通道電流在最大舒張電位時可用。Cav3.1介導的鈣通道電流穩態激活和失活曲線的相對位置不能表明有顯著的窗口電流分量[16]。此外,T型鈣通道已經在SAN、AVN和浦肯野纖維這3個心臟節律中心中被觀察到[21],表明T型鈣通道可能與舒張期去極化的產生有關。
Chen等[15]通過靶向滅活Cav3.2和Cav3.1的轉基因小鼠闡明T型鈣通道異構體在心臟起搏和脈沖傳導中的作用。與野生型小鼠相比,通過同源基因靶向技術構建的Cav3.2缺陷小鼠,其心率和心電圖波形無顯著差異,表明T型鈣通道中的Cav3.2不能顯著促進心臟自動性的產生和傳導。與Cav3.2缺陷小鼠相反,小鼠的Cav3.1基因失活導致SAN傳導速率中度降低和房室間隔傳導延長。Cav3.1通道相關的通道病強調了鈣通道在心臟自動性的產生和調節中的重要性[22]。這些研究說明在心臟自律性的產生和傳導中,T型鈣通道的Cav3.1起主要作用。
3.1.2 T型鈣通道缺陷導致先天性心臟傳導阻滯(congenital heart block,CHB)發生CHB是一種被動獲得的自身免疫病,發生于患有風濕性疾病的母親的妊娠期,并與母親抗Ro/SSA和抗La/SSB抗體有關。該病的特征是房室傳導阻滯,可在妊娠16~25周的胎兒中檢測到[23]。Cav3.1缺陷小鼠出現SAN傳導速率中度降低和房室間隔傳導延長,這是CHB的一種表型[24]。這提示Cav3.1在CHB的發展過程中可能與抗Ro抗體陽性IgG發生交叉反應[25]。Strandberg等[26]觀察到人類孕18~22.6周胎兒心臟AVN中T型鈣通道Cav3.1的mRNA表達與心尖區存在差異。實驗結果表明CHB胎兒心肌細胞表面可表達Cav3.1,并影響妊娠的孕婦血清對α1G蛋白的反應性。CHB孕婦血清對α1G的反應顯著高于對照組,反應性的表位被定位到一個p305的肽段(對應于連接跨膜片段S5-S6的細胞外環aa305-319)。單細胞膜片鉗電生理學實驗也證實了CHB母體血清會不可逆地降低小鼠SAN細胞T型Ca2+電流。因此CHB母體血清抗體容易靶向人類胎兒心肌細胞中T型鈣通道Cav3.1的外表位。
自身抗體在Cav3.1[26]和Cav1.3[27]通道α1亞基上存在結合位點。在這2個通道中,結合位點都位于連接α1亞基的S5和S6域胞外環路,靠近通道孔。Cav1.3和Cav3.1通道中結合位點的定位與觀察到急性灌注自身抗體能迅速抑制Cav1.3介導的L型鈣通道和Cav3.1介導的T型鈣通道的結果一致。人類胎兒心臟CHB的發生可能涉及2個階段:在第一階段,母體自身抗體結合α1亞基抑制Cav1.3和Cav3.1通道;在第二階段,VGCC在肌細胞中內化,同時這也是不可逆的階段。此外,有研究也提出長期暴露于母體自身抗體和通道內化可引發肌細胞死亡而導致CHB的產生,并在患者死后的心臟中檢測到SAN和AVN的纖維化和鈣化[27]。
L型Cav1.3和T型Cav3.1鈣通道在心臟自動性的產生中起重要作用。自21世紀以來,轉基因小鼠已經幫助確定了離子通道亞型在起搏器活性中的作用[16]。重要的是,這些小鼠系再現了由于Cav1.3通道突變和Cav3.1通道自身免疫功能喪失而導致的某些形式的遺傳性SAN病變。
3.1.3 不同亞型T型鈣通道對心肌肥大的作用不同心臟能夠通過增加肌肉的質量從而應對壓力的變化,但長期處于壓力超負荷下心臟會出現一種常見的適應性反應,稱為心肌肥大,其持續進展會導致心力衰竭[28-29]。心肌肥大的特征包括細胞大小改變、肌動蛋白重組、蛋白質合成增加和胎兒基因重新表達。在肥大心臟中重新表達的胎兒基因包括心房鈉尿肽、腦鈉肽、收縮蛋白的胎兒亞型(α-骨骼肌肌動蛋白和β-肌球蛋白重鏈),以及胎兒心臟中表達的離子通道,如超極化激活的環核苷酸門控通道和T型鈣通道[30]。長期心肌肥大可能會導致心力衰竭和死亡,病理性心肌肥大通常與心臟的結構和功能重塑有關,其中參與心臟調節的鈣通道發揮重要的作用[31]。
目前不同亞型的T型鈣通道在心肌肥大中的作用不同。Chiang等[32]提出,Cav3.2通過激活鈣調磷酸酶(calcineurin)/活化T細胞核因子(nuclear factor of activated T cells,NFAT)途徑參與心肌肥大的發病機制。在缺乏Cav3.2的小鼠中,壓力超負荷誘導的肥大受到嚴重抑制,但在缺乏Cav3.1基因的小鼠中卻沒有明顯表現。這一結果說明Cav3.2通過激活calcineurin-NFAT信號級聯調節病理性心肌肥大,并對心肌肥大的產生起重要作用。而Nakayama等[33]卻指出Cav3.1能夠對抗心肌肥大的發生和發展。通過對具有可誘導表達Cav3.1的心臟特異性轉基因小鼠與Cav3.1缺陷小鼠進行壓力超負荷、異丙腎上腺素輸注和運動誘導等,結果顯示Cav3.1轉基因小鼠對心肌肥大具有抵抗力,沒有產生心臟的病理表現,而在Cav3.1缺陷小鼠中卻表現出增強的肥大反應。在機制上,Cav3.1保護心臟是通過調節一氧化氮合酶3(nitric oxide synthase 3,NOS3)實現的,NOS3是一種鈣激活的信號效應器,已知可改變心臟肥大反應。Cav3.1與NOS3相互作用,后者在壓力超負荷后增加了Cav3.1轉基因小鼠心臟中環鳥苷酸(cyclic guanosine monophosphate,cGMP)依賴性蛋白激酶I型的活性,從而實現Cav3.1對抗心肌肥大的作用[34]。這些實驗結果表明Cav3.1對心肌肥大有抑制作用,而Cav3.2對心肌肥大的激活具有重要意義,這2種不同亞型的T型鈣通道作用于心肌,對心肌肥大的發生起到調節和平衡作用。
3.2 T型鈣通道參與抑制血管過度收縮除心肌細胞外,血管平滑肌細胞亦可表達除Cav1.2外的其他VGCC。鈣通道可使血管感知和響應生理刺激,進而調節動脈張力,維持血管功能。其中,T型鈣通道受到了廣泛關注[35]。Cav3.2在超極化的血管平滑肌細胞局部微結構域信號傳遞中發揮重要作用,通過調控動脈張力的反饋抑制,引起阻力動脈的擴張[36]。
T型Cav3.2通道介導的Ca2+內流刺激RyR的細胞質結構域,以Ca2+觸發的形式促進肌漿網釋放Ca2+,引起K+外流和大電導鈣激活鉀通道(large-conductance calcium-activated potassium channels,BKCa)的激活,從而避免血管過度收縮。為了使這個信號通路正常的發揮作用,T型Cav3.2通道必須與RyR緊密地排列在一起[37]。越來越多研究表明,年齡是影響血管平滑肌功能的關鍵因素,人們逐漸認識到鈣通道信號會隨著衰老而改變,而且會影響血管的功能。Fan等[38]利用不同年齡(4和12月齡)小鼠血管平滑肌細胞的高空間分辨率共聚焦Ca2+成像,探究衰老對血管平滑肌細胞中的Cav3.2-RyR-BKCa信號傳導通路的影響。該研究表明,衰老破壞了Cav3.2與RyR的偶聯,通過蛋白表達分析和電鏡觀察結果顯示,這種作用可能是由于與年齡相關的Cav3.2蛋白表達減少和血管平滑肌細胞微囊結構變化引起的。此外,Georgeon-Chartier等[39]報道了衰老小鼠血管平滑肌細胞RyR介導的Ca2+信號減少,RyR2表達下降,血管收縮能力下降。這些結果表明,衰老可通過破壞Cav3.2-RyR-BKCa信號通路導致血管收縮能力下降。
4 T型鈣通道的調節劑
4.1 血管緊張素受體拮抗劑抑制T型鈣通道表達眾所周知,腎素-血管緊張素系統的激活可以靶向血管平滑肌細胞,從而升高血壓,導致高血壓,而且該系統同樣也與心肌肥大和致死性心律失常有關[40]。該系統的主要分子血管緊張素II已成為影響許多細胞類型和器官功能的關鍵激素,并在心律失常和心房顫動的發展中起著重要作用[41]。
T型鈣通道在心律失常中起重要作用,血管緊張素II 1型(angiotensin II type 1,AT1)受體拮抗劑作為抗心律失常藥物發揮作用。心臟的電重構通常與細胞內Ca2+過載有關,T型鈣通道能將Ca2+帶入心肌細胞,提示T型鈣通道在心臟自律性中發揮生理作用。在病理狀態下,T型鈣通道在心肌細胞中重新表達[42]。心臟電生理特性的變化與包括T型鈣通道在內的多種離子通道基因表達的變化有關,這些變化容易導致心律失常的發生[43]。但是T型鈣通道如何參與血管緊張素II介導的心律失常的發生,仍需進一步的實驗研究。Morishima等[44]研究AT1受體拮抗劑替米沙坦對大鼠新生心肌細胞T型鈣通道的表達和心肌收縮力的轉錄調節的影響。用替米沙坦和血管緊張素II培養心肌細胞24 h后測量T型Ca2+電流及通道。其結果顯示,長期應用血管緊張素II(24 h)后,心肌細胞的T型Ca2+電流密度顯著增加,并伴有細胞外信號調節激酶1/2(extracellular signal-regulated kinase 1/2,ERK1/2)和p38絲裂原活化蛋白激酶(p38 mitogen-activated protein kinase,p38 MAPK)磷酸化。而使用替米沙坦則降低了Cav3.1和Cav3.2的mRNA表達以及T型Ca2+電流。此外,在缺乏血管緊張素II的情況下,替米沙坦降低了p38 MAPK的基礎磷酸化水平,但沒有降低ERK1/2的水平。上述結果表明,替米沙坦可通過不依賴激動劑的方式抑制p38 MAPK活性并減弱T型鈣通道的表達,從而起到抗心律失常的作用(圖1)。
4.2 腎上腺素增強T型鈣通道表達心率調節是通過SAN的自動性和傳導率的改變發生的,當交感神經受到刺激時,交感神經/β-腎上腺素能系統興奮,釋放兒茶酚胺,與β-腎上腺素能受體結合并激活環腺苷酸(cyclic adenosine monophosphate,cAMP)/蛋白激酶A(protein kinase A,PKA)信號[45]。由于β-腎上腺素能系統對心率調節至關重要,而Cav3.1參與心率產生,因此可以驗證β-腎上腺素/PKA系統是否參與對T型鈣離子通道的調節。此外,cAMP依賴性PKA對T型鈣通道的調控一直存在爭議,這可能是由于實驗條件、細胞類型的差異以及特定亞型的存在[46]。
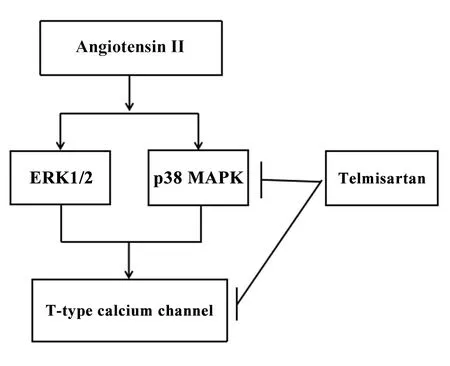
Figure 1.The role of angiotensin and T-type calcium channel.The T-type Ca2+current density in cardiomyocytes was increased after angiotensin II treatment via activating extracellular signal-regulated kinase 1/2(ERK1/2)and p38 mitogen-activated protein kinase(p38 MAPK).In contrast,telmisartan decreased the density of T-type Ca2+current in a dose-dependent manner by inhibiting the basal phosphorylation level of p38 MAPK in the absence of angiotensin II.圖1 血管緊張素與T型鈣通道的作用
Li等[47]通過Cav3.1雙轉基因小鼠心室肌細胞,以及野生型小鼠、Cav3.1敲除小鼠和Cav3.2敲除小鼠SAN細胞的研究,觀察到β-腎上腺素能受體激動劑異丙腎上腺素能夠增強過表達Cav3.1通道的活性。這種作用是由腺苷酸環化酶(adenylate cyclase,AC)/cAMP/PKA系統介導的,因為cAMP能夠表現異丙腎上腺素的作用效果,而PKA抑制劑H89可以消除異丙腎上腺素對Cav3.1的作用。由此說明cAMP/PKA通路介導β-腎上腺素能受體刺激而增強Cav3.1活性。之后,Li等[48]提出β-腎上腺素能受體通過刺激T型鈣通道活性上調,誘導心率升高和房室傳導加速。對小鼠注射異丙腎上腺素,其EC50在Cav3.1轉基因小鼠的心率應答中較在Cav3.1基因敲除小鼠中低。而在異丙腎上腺素使小鼠心率應答的占比中,Cav3.1轉基因小鼠高于Cav3.1基因敲除小鼠。而且Cav3.1基因敲除小鼠的T型Ca2+電流消失,Cav3.1轉基因小鼠中的T型Ca2+電流增強。這說明小鼠SAN中的T型Ca2+電流是由Cav3.1介導的,β-腎上腺素能受體刺激心率上調和房室傳導加速的作用在KO小鼠體內減弱,而在Cav3.1轉基因小鼠體內加強;Cav3.1的表達影響SAN細胞自律性,而且其表達通過β-腎上腺素能受體刺激調節。這些研究說明β-腎上腺素能受體刺激Cav3.1活性增強在小鼠心率調節中發揮重要作用,β-腎上腺素能系統調控Cav3.1異常可能會導致心臟自律性異常(圖2)。

Figure 2.The role of adrenaline and T-type calcium channel.βadrenergic receptor agonist increased the expression of Cav3.1 by activating adenylate cyclase(AC)-cyclic adenosine monophosphate(cAMP)-protein kinase A(PKA)signaling pathway,and H89,a PKA inhibitor,could inhibit the phosphorylation of PKA and reduced the T-type Ca2+current.圖2 腎上腺素與T型鈣通道的作用
4.3 醛固酮間接調控T型鈣通道表達鹽皮質激素,尤其是醛固酮,與心肌肥大和心力衰竭的發生密切相關。心臟中鹽皮質激素受體(mineralocorticoid receptor,MR)的激活是心血管疾病發生的危險因素。MR激活會導致包括鈣通道在內的許多離子通道的上調,發生心臟肥大和心律失常[49]。Messaoudi等[50]對心肌細胞靶向性MR過度表達的小鼠及其對照組給予1周低劑量醛固酮治療,確定醛固酮是否能在體內激活心肌細胞MR。通過分析心臟轉錄組,觀察到心肌細胞中存在醛固酮調節的基因,并激活心肌細胞MR。由于T型鈣通道內不包含任何鹽皮質激素反應元件(mineralocorticoid response element,MRE;與MR錨定的結構域一致的核苷酸序列),醛固酮對T型鈣通道表達的控制可能是間接的[51],可能是由于心肌細胞MR替代了MRE的作用。
心血管疾病與微小RNA(microRNA,miRNA,miR)的調節密切相關[52]。miRNA調節大量信號通路,參與細胞間的信息交流,并參與心血管系統的生理病理進程,如調控心肌細胞分化、促進血管新生、維持內皮細胞穩定性及抑制內皮遷移和增殖等。Rossier[53]提出miR-204在MR下游發揮作用以控制T型鈣通道表達。Koyama等[51]假定一種特定的miRNA在MR激活與T型鈣通道表達和心肌細胞搏動頻率的調節中起到聯系作用。通過一項篩選實驗發現,在醛固酮刺激離體乳鼠心肌細胞后,miR-204是主要上調的miRNA之一。當miR-204在分離的心肌細胞中過度表達時,其自發搏動頻率在24 h后顯著增加,與受到醛固酮刺激時表現相同,編碼T型鈣通道的mRNA增加。同時,miR-204過度表達后,T型Ca2+電流顯著增加。當抑制miR-204的表達時,會消除醛固酮誘導Cav3.1和Cav3.2的mRNA表達增加,同時T型Ca2+電流也會減少。此外,Koyama等[51]還觀察到醛固酮和miR-204的過度表達降低了已知的Cav3轉錄抑制因子——神經元限制性沉默因子(neuron-restrictive silence factor,NRSF)。由此可以說明,醛固酮對T型鈣通道的調控是間接實現的,醛固酮對心肌細胞MR的刺激會導致miR-204的表達,而miR-204的表達促進了分離的大鼠心室肌細胞中T型鈣通道的表達,增加了細胞自發收縮的頻率,這是通過抑制NRSF蛋白實現的(圖3)。

Figure 3.The role of mineralocorticoid and T-type calcium channel.The stimulation of mineralocorticoid receptor(MR)by aldosterone could increase T-type Ca2+current via microRNA-204(miR-204).Neuron-restrictive silencing factor(NRSF)inhibited this pathway by lessening the level of miR-204 and T-Type calcium channel.圖3 鹽皮質激素與T型鈣通道的作用
5 結語
VGCC不僅能夠改變細胞電位,還能參與信號轉導。不同于其它高電壓激活的鈣通道,T型鈣通道以其低電壓激活的特性在心臟血管的生理活動中發揮著重要的作用,參與心臟自律性的產生和傳導,其功能缺陷可導致先天性心臟傳導阻滯的發生。不同亞型的T型鈣通道對心肌肥大作用不同作用。除心肌本身外,T型鈣通道還可參與抑制血管過度收縮,進而維持正常血管收縮舒張功能。近年來對于T型鈣通道在電生理傳導通路作用及分子調控機制仍是研究熱點問題,雖然已證實其受腎上腺素、醛固酮等多種激素的直接或間接調控,但其他調控因素及相應的臨床轉化應用仍需進一步的探索。
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
東方少年·布老虎畫刊(2023年8期)2023-08-01 15:45:12
科學大眾(2021年6期)2021-07-20 07:42:44
科學(2020年3期)2020-11-26 08:18:30
學苑創造·A版(2020年9期)2020-10-13 09:41:02
娃娃樂園·綜合智能(2019年3期)2019-04-03 09:17:36
中成藥(2018年2期)2018-05-09 07:19:34
小學生學習指導(低年級)(2017年10期)2017-10-10 01:00:05
湖北師范大學學報(自然科學版)(2015年2期)2016-01-10 08:41:55
云南中醫學院學報(2014年3期)2014-07-31 18:57:34
