過表達miR-202-5p 通過抑制PCSK9 減輕阿爾茨海默病神經損傷
2022-12-24 02:41:56徐沛沛趙樹華王江波姚先麗白
中國比較醫學雜志 2022年10期
關鍵詞:模型
徐沛沛趙樹華王江波姚先麗白 金*
(1.鄭州大學附屬鄭州中心醫院康復醫學科,鄭州 450001;2.河南中醫藥大學附屬鄭州人民醫院康復醫學科,鄭州 450014)
阿爾茨海默癥(Alzheimer’s disease,AD)多發于老年人群,是誘導老年人死亡的第五大疾病[1]。過往研究發現,隨著年齡及衰老的發展,β-淀粉樣蛋白(amyloid β,Aβ)大量積累引起的神經炎性反應是導致AD 患者神經系統損傷及記憶喪失、認知障礙及行為改變的主要原因[2]。 近來有研究發現,腦內膽固醇失衡,是導致AD 患者Aβ 代謝異常及神經炎癥發生的關鍵因素之一[3]。 文獻報道發現,具有監管膽固醇穩態的重要調節因子-前蛋白轉化酶枯草溶菌素9(proprotein convertase subtilisin/kexin type 9,PCSK9)可在AD 患者的腦脊液中呈異常高表達[4],且PCSK9的高表達一方面可通過抑制神經元攝取膽固醇而阻斷神經元發育再生[5],另一方面可通過影響β 位點APP 剪切酶1(β-site APP cleaving enzyme 1,BACE1)表達來引起Aβ 加工及代謝異常,而導致Aβ 積聚及神經炎癥反應的發生[6]。 但引起AD 患者PCSK9 異常變化的基因調控機制還不甚清楚。
miRNA 是參與AD 神經系統病變的重要調控因子,大量研究發現,AD 患者Aβ 積聚,與miRNA 的異常表達關系密切[7]。 已有文獻顯示,miR-202-5p可通過靶向抑制淀粉樣前體蛋白(amyloid precursor protein,APP)的3’UTR 端,而降低AD 患者Aβ 沉積引起的神經系統損傷[8]。 但AD 患者PCSK9 表達的升高是否與miR-202-5p 表達的異常有關還未見報導。 本研究建立AD 體內外模型,干預miR-202-5p 及PCSK9 表達后,對此進行驗證探究,以期為AD 的基因靶向治療提供新的思路。
1 材料和方法
1.1 實驗動物
清潔級SD 雄性大鼠50 只,體重260~280 g,由中國醫學科學院醫學實驗動物研究所提供[SCXK(京)2021-0004]。 所有大鼠均飼養在鄭州大學(藥物研究院)[SYXK(豫)2018-0004],飼養溫度為22℃~25℃,濕度控制在55%左右。 無菌手術在鄭州大學(藥物研究院)進行.本實驗經鄭州大學附屬鄭州中心醫院動物倫理委員會批準(20210617-0012),并按實驗動物使用的3R 原則給予人道的關懷。
1.2 主要試劑與儀器
小鼠腦皮質神經元(Sci-L-0956,上海圻明生物科技有限公司);PCSK9 抑制劑-Aliro(YSRIBIOC4745,上海研生實業有限公司);Aβ1-42(A4558,美國Sigma 公司,使用前,將Aβ1-42 肽以200 μmol/L 濃度溶解于無菌雙蒸水中,37℃靜置24 h,14000 r/min、4℃離心10 min 制成可溶性Aβ1-42 寡聚體備用);Aβ42、Aβ40 ELISA 試劑盒(JH-R30135、JHR30130,上海繼和生物科技有限公司);PCSK9 ELISA 試劑盒(EK-M28850,上海酶研生物科技有限公司);腫瘤壞死因子-α(TNF-α)、白細胞介素-1β(IL-1β)、磷酸化Tau 蛋白(P-tau)、總膽固醇(TC)ELISA 試 劑 盒 ( FN-AQ-M0183、 E-R-0067、1529384953、EK-R38996,武漢菲恩生物科技有限公司、深圳海思安生物技術有限公司、上海江萊生物科技有限公司、上海酶研生物科技有限公司);PCSK9、負責神經元膽固醇攝取相關因子-LDL 受體相關蛋白1(LRP-1)、載脂蛋白E(ApoE)及APP、BACE1 等兔抗大鼠抗體均購自英國abcam 公司。FC 酶標儀購自ThermoFisher;BX43 光學顯微鏡購自日本奧林巴斯;Dounce 組織勻漿器購自海德創業(北京)生物科技有限公司。
1.3 實驗方法
1.3.1 模型建立及分組
取大鼠40 只,參照文獻[9]方法,將大鼠麻醉后,經側腦室注射5 μL 濃度為80 μmoL/L 的Aβ1-42,2 d 1 次,連續8 d,建立大鼠AD 模型,用水迷宮試驗檢測大鼠學習記憶能力,若大鼠出現學習記憶能力下降現象,視為造模成功。 將造模成功大鼠按隨機數字表法分模型組、ago-miR-202-5p 組、ago-NC組、PCSK9 抑制劑組,每組10 只,另取10 只大鼠側腦室注射5 μL 無菌雙蒸水,相同時間后,作為正常對照組。 各組于造模成功后開始給藥,ago-miR-202-5p 組及ago-NC 組經側腦室注射5 μL 濃度為25 μmoL/L 的miR-202-5p 激動劑(ago-miR-202-5p)及陰性對照(ago-NC)進行干預治療,3 d 1 次,連續15 d;PCSK9 抑制劑-Aliro 參照文獻[10]經側腦室注射16 mg/kg 的PCSK9 抑制劑Aliro,3 d 1 次,連續15 d。 正常對照組及模型組經側腦室注射等量雙蒸水。
1.3.2 大鼠空間記憶及學習能力檢測
制備水迷宮試驗平臺(直徑為150 cm 的圓形水池,水深60 cm,水溫(23±2)℃,水池分為4 個象限,第一象限內含距水面1~2 cm 的逃生平臺,直徑14 cm,高29 cm)。 將大鼠面朝池壁從第4 象限放入水中,120 s 后未找到平臺的引導其找到平臺,并讓其在平臺上停留10 s,進行連續訓練5 d,每天訓練4次,第6 天用攝像頭記錄大鼠自主找到逃生平臺的時間,即逃避潛伏期,逃逸潛伏期越短,預示其空間記憶能力越強。 撤去水下逃生平臺,將大鼠從第4象限面朝池壁投入池中,并讓其自由游泳120 s,攝像頭記錄統計大鼠120 s 內穿越原平臺位置的次數,穿越原平臺次數越高預示其空間學習能力越好。
1.3.3 ELISA 法檢測腦脊液及腦皮層組織相關因子
各組大鼠,斷頭取腦,解剖取腦脊液2 mL,按ELISA 試劑盒方法檢測PCSK9、Aβ42、Aβ40 及TC水平;剝取完整腦皮層組織,分成兩部分,一部分置于4%多聚甲醛中固定24 h 備用,另一部分剪取約1 g 粉碎研磨后取勻漿液,按ELISA 試劑盒方法檢測TNF-α、IL-1β、P-tau 水平,剩余腦皮層組織置于-80℃保存。
1.3.4 尼氏及神經元核抗原(NeuN)免疫熒光染色法檢測腦皮層組織神經元形態及數目變化
將4%多聚甲醛中固定的腦皮層組織,制成厚為5 μm 的石蠟切片,脫蠟、透化及抗原修復處理后,按尼氏染色液說明書方法染色后,于光鏡下觀察神經元形態變化,滴加1 ∶500 的NeuN 一抗孵育過夜后,用0.5%的驢抗兔二抗IgG 避光孵育處理10 min 后,DAPI 核染10 min,晾干及封片后,光鏡下觀察,Image Pro-Plus 6.0 軟件計算每視野下單位面積下NeuN 陽性染色神經元數目。
1.3.5 qRT-PCR 法檢測腦皮層組織miR-202-5p表達
粉碎腦皮層組織,提取RNA,逆轉錄得到cDNA,行PCR 反應(反應體系:cDNA 1 μL;SYBR GREEN 反 應 液5 μL,PCR 引 物 各0.8 μL,H2O 3 μL,反應條件:90℃預變性60 s、1 個循環,94℃變性10 s、65℃退火60 s、45 個循環)。 miR-202-5p( 5 ’-GTCACATGCTGGGTCCAACAAA-3 ’, 5 ’-GCGTCCTATGCACTC-3’)以U6 為內參用2-ΔΔCt法計算基因相對表達水平,引物序列由大連寶生生物公司合成。
1.3.6 Western blot 法檢測腦皮層組織相關蛋白表達
粉碎腦皮層組織,組織勻漿器勻漿得勻漿液,提取蛋白,BCA 法測定蛋白總濃度,制備濃縮膠及分離膠,取60 μg 蛋白行電泳和轉膜反應,加入1 ∶900 的一抗PCSK9、LRP-1、ApoE、APP、BACE1 抗體,及1 ∶1 200 的內參β-actin 抗體,4℃搖床孵育過夜,HRP 羊抗兔二抗室溫孵育40 min,化學發光法液顯色后,用以Image J 軟件分析條帶相對灰度值。
1.3.7 雙熒光素酶報告試驗驗證miR-202-5p 與PCSK9 的靶向關系
starbase 網站預測miR-202-5p 和PCSK9 的結合位點。 建立含有miR-202-5p 結合位點的PCSK9-3’UTR 野生型(WT)和PCSK9-3’UTR 突變型(MUT)片段,克隆至熒光素酶報告質粒中,Lipofectamine 2000 轉 染 試 劑 將PCSK9-3’ UTR-WT、PCSK9-3’UTR-MUT 熒光素酶報告質粒,分別與mimic NC 或miR-202-5p mimic 共轉染至293 T 細胞中,48 h 后檢測熒光素酶活性。
1.3.8 AD 細胞模型共轉染miR-202-5p mimic 及PCSK9 過表達質粒(pcDNA-PCSK9)對皮質神經元損傷的影響
取小鼠原代皮質神經元凍存管,37℃水浴復蘇后,取適量加入DMEM 培養基(含5%胎牛血清)中進行傳代培養及計數。 取對數期生長良好的神經元,按1×108個/孔的密度接種于6 孔板內,設置為空白組、Aβ 誘導組(參照文獻[11]加入終濃度為1 μmol/L 凝聚態的Aβ1-42 培養1 h 作為AD 細胞模型)、miR-202-5p mimic 組(在Aβ 誘導組基礎上轉染miR-202-5p mimic)、mimic-NC 組(在Aβ 誘導組基礎上轉染miR-202-5p mimic 陰性對照載體-mimic-NC)、miR-202-5p mimic + pcDNA-PCSK9 組(在Aβ 誘導組基礎上共轉染miR-202-5p mimic 及pcDNA-PCSK9)、miR-202-5p mimic+pcDNA-NC 組(在Aβ 誘導組基礎上共轉染miR-202-5p mimic 及pcDNA-PCSK9 陰性對照載體-pcDNA-NC),每組設置6 個復孔。 空白組加入培養基正常培養;Lipofectamine 2000 轉染試劑進行轉染,各組細胞培養48 h 后,取細胞上清液,用ELISA 法檢測PCSK9、TC、 TNF-α、 IL-1β、 P-tau 水 平; 取 細 胞 按Honchest33258 染色液說明書方法,染色后于顯微鏡下觀察細胞核損傷狀況;按1.3.5 及1.3.6 方法測基因及相關蛋白表達。
1.4 統計學方法
以SPSS 22.0 軟件對實驗數據進行統計分析,計量資料以平均數±標準差(±s)表示,多組間比較進行單因素方差分析,進一步兩組間比較行snk-q檢驗,P<0.05 表示差異有統計學意義。
2 結果
2.1 過表達miR-202-5p 或抑制PCSK9 對大鼠腦皮層組織miR-202-5p 表達的影響
與正常對照組(1.09±0.10)相比,模型組大鼠腦皮層組織中miR-202-5p 表達(0.14±0.10)降低(P<0.05)。 與模型組相比,ago-miR-202-5p 組大鼠腦皮層組織中miR-202-5p 表達(0.81±0.12)升高(P<0.05);PCSK9 抑制劑組(0.11±0.11)、mimic-NC 組(0.12±0.10)大鼠腦皮層組織miR-202-5p 表達與模型組相比差異不顯著(P>0.05)。
2.2 過表達miR-202-5p 或抑制PCSK9 對AD 大鼠空間學習記憶能力的影響
與正常對照組相比,模型組大鼠逃避潛伏期延長,穿越平臺次數減少(P<0.05),預示其空間記憶及學習能力降低。 與模型組相比,ago-miR-202-5p 組及PCSK9 抑制劑組大鼠逃避潛伏期縮短,穿越平臺次數升高(P<0.05),且ago-miR-202-5p 組對上述指標的改善效果,好于PCSK9 抑制劑組(P<0.05)。 ago-NC組與模型組相比,差異不顯著(P>0.05),見表1。
表1 大鼠平均逃避潛伏期、穿越平臺次數(±s,n=10)Table1Averageescapelatencyandtimesof crossing the platform in rats
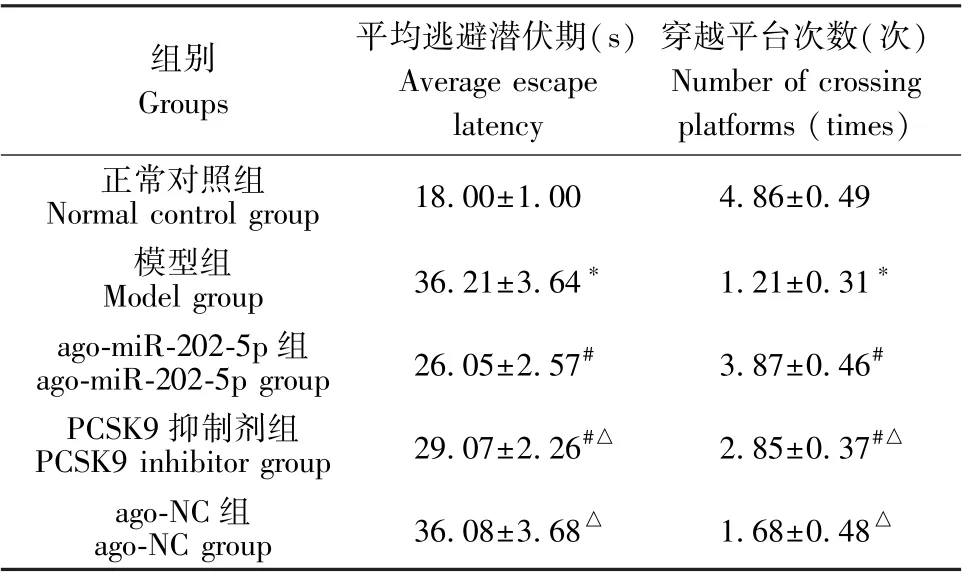
表1 大鼠平均逃避潛伏期、穿越平臺次數(±s,n=10)Table1Averageescapelatencyandtimesof crossing the platform in rats
注:與正常對照組相比,*P<0.05;與模型組相比,#P<0.05;與agomiR-202-5p 組相比,△P<0.05。Note. Compared with the normal control group,*P<0.05. Compared with the model group,#P<0.05. Compared with the ago-miR-202-5p group,△P<0.05.
G組ro別ups平均Av逃er l避aagt e e潛n ec伏syc a期pe(s)穿Npu越l a m tf平boer臺mr s o次f(c數trioms(es次si n)g)Norm正al常 co對nt照rol組 group 18.00±1.00 4.86±0.49 Mo模de型l g組roup 36.21±3.64* 1.21±0.31*aga og-omm iRiR-2-0220-25-p5 pg r組oup 26.05±2.57# 3.87±0.46#PCPSCK S9K i9n h抑i b制ito劑r g組roup 29.07±2.26#△ 2.85±0.37#△aga og-oNNCC g r組oup 36.08±3.68△ 1.68±0.48△
2.3 過表達miR-202-5p 或抑制PCSK9 對AD 大鼠腦皮層神經元形態變化的影響
正常對照組大鼠腦皮層神經元細胞結構正常,層次清晰,其數目為每平方毫米(55.50±5.00)個。模型組大鼠神經元細胞體積變小,排列紊亂、核固縮且染色較深、胞體拉長收縮呈多角形,NeuN 陽性染色的神經元細胞數目為每平方毫米(30.05±3.00)個,與正常對照組相比減少(P<0.05)。 agomiR-202-5p 組及PCSK9 抑制劑組大鼠腦皮層神經元核固縮、胞體拉長收縮現象緩解,NeuN 陽性染色的神經元細胞數目為每平方毫米(49.02±4.10)個、(40.00±4.00)個,與模型組相比增多,且ago-miR-202-5p 組對神經元損傷的改善作用優于PCSK9 組(P<0.05)。 ago-NC 組神經元細胞數目為每平方毫米(30.20±3.02)個,與模型組相比,神經元形態相近,數目差異不顯著(P>0.05),見圖1。
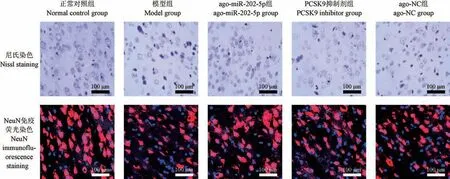
圖1 大鼠腦皮層組織尼氏染色及NeuN 免疫熒光染色圖Figure 1 Nissl staining and NeuN immunofluorescence staining of rat cerebral cortex
2.4 過表達miR-202-5p 或抑制PCSK9 對AD 大鼠腦脊液中PCSK9、Aβ42、Aβ40、TC 水平的影響
與正常對照組相比,模型組大鼠腦脊液中PCSK9、Aβ42、Aβ40、TC 水平升高(P<0.05)。 與模型組相比,ago-miR-202-5p 組及PCSK9 抑制劑組大鼠腦脊液中PCSK9、Aβ42、Aβ40、TC 水平降低(P<0.05),且ago-miR-202-5p 組對上述指標的改善效果好于PCSK9 抑制劑組(P< 0.05)。ago-NC 組與模型組相比,上述指標差異不顯著(P>0.05),見圖2。

圖2 大鼠腦脊液中PCSK9、Aβ42、Aβ40、TC 水平比較Note. Compared with the normal control group,*P<0.05. Compared with the model group,#P<0.05. Compared with the ago-miR-202-5p group,△P<0.05.Figure 2 Comparison of PCSK9, Aβ42, Aβ40 and TC levels in rat cerebrospinal fluid
2.5 過表達miR-202-5p 或抑制PCSK9 對AD 大鼠腦皮層組織中TNF-α、IL-1β、P-tau 水平的影響
與正常對照組相比,模型組大鼠腦皮層組織中TNF-α、IL-1β、P-tau 水平升高(P<0.05)。 與模型組相比,ago-miR-202-5p 組及PCSK9 抑制劑組大鼠上述指標水平降低(P<0.05),且ago-miR-202-5p 組對上述指標的改善效果好于PCSK9 抑制劑組(P<0.05)。 ago-NC 組與模型組相比,上述指標差異不顯著(P>0.05),見圖3。
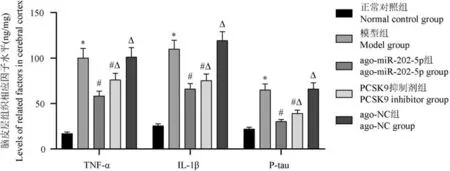
圖3 大鼠腦皮層組織中TNF-α、IL-1β、P-tau 水平比較Note. Compared with the normal control group,*P<0.05. Compared with the model group,#P<0.05. Compared with the ago-miR-202-5p group,△P<0.05.Figure 3 Comparison of TNF-α, IL-1β and P-tau levels in rat cerebral cortex
2.6 過表達miR-202-5p 或抑制PCSK9 對AD 大鼠腦皮層組織PCSK9 及下游LRP-1、ApoE、APP、BACE1 蛋白表達的影響
與正常對照組相比,模型組大鼠腦皮層組織中LRP-1、ApoE 表達降低,PCSK9、APP、BACE1 蛋白表達升高(P<0.05)。 與模型組相比,ago-miR-202-5p 組及PCSK9 抑制劑組大鼠腦皮層組織中LRP-1、ApoE 表達升高,PCSK9、APP、BACE1 蛋白表達降低(P<0.05),且ago-miR-202-5p 組對上述指標的改善效果好于PCSK9 抑制劑組(P<0.05)。 ago-NC 組與模型組相比,上述指標差異不顯著(P>0.05),見圖4。
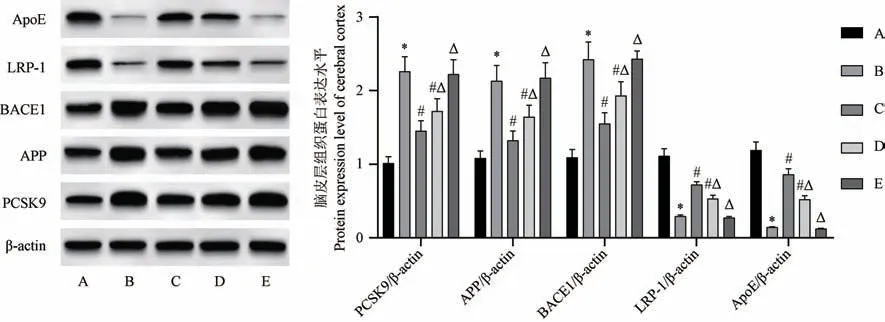
圖4 大鼠腦皮層組織蛋白表達水平比較Note. A, Normal control group. B, Model group. C, ago-miR-202-5p group. D, PCSK9 inhibitor group. E, ago-NC group. Compared with the normal control group,*P<0.05. Compared with the model group,#P<0.05. Compared with the ago-miR-202-5p group,△P<0.05.Figure 4 Comparison of protein expression levels in rat cerebral cortex
2.7 miR-202-5p 對PCSK9 靶向調控的影響
starbase 網站預測miR-202-5p 與PCSK9 存在結合位點。 雙熒光素酶試驗發現,共轉染miR-202-5p mimic 和無突變的PCSK9-WT 載體,可使293 T 細胞中熒光素酶活性顯著下降(0.98±0.07 vs 0.34±0.04,P<0.01),而共轉染miR-202-5p mimic 和發生突變的PCSK9-MUT 載體對熒光素酶活性無顯著作用(0.97±0.07 vs 0.93±0.08,P>0.05),見圖5。

圖5 miR-202-5p 和PCSK9 的靶向關系Figure 5 Targeting relationship between miR-202-5p and PCSK9
2.8 共轉染miR-202-5p 及pcDNA-PCSK9 對AD模型皮質神經元細胞損傷及相應水平的影響
Honchest 33258 染色可見,空白組神經元細胞核形態正常;Aβ 誘導組神經元細胞核不規則、核膜破裂、核質固縮、核仁碎裂呈顆粒狀;miR-202-5p mimic 組及miR-202-5p mimic+pcDNA-NC 組神經元細胞胞膜較完整,細胞核固縮及損傷較Aβ 誘導組減輕;miR-202-5p mimic+pcDNA-PCSK9 組神經元細胞損傷較miR-202-5p mimic 組加重;mimic-NC 組細胞損傷與模型組相近,見圖6。

圖6 腦皮質神經元Honchest33258 染色圖Figure 6 Honchest33258 staining of cerebral cortical neurons
與空白組相比,Aβ 誘導組細胞上清液中PCSK9、TC、TNF-α、P-tau 水平升高(P<0.05)。 Aβ誘導組相比, miR-202-5p mimic 組、 miR-202-5p mimic+pcDNA-NC 組、miR-202-5p mimic+pcDNAPCSK9 組細胞上清液中上述水平降低(P<0.05)。與miR-202-5p mimic 組相比,miR-202-5p mimic+pcDNA-PCSK9 組細胞上清液中各指標升高(P<0.05)。 mimic-NC 組與模型組相比,miR-202-5p mimic 組與miR-202-5p mimic+pcDNA-NC 組相比,上述指標差異不顯著(P>0.05),見圖7。

圖7 細胞上清液中PCSK9、TC、TNF-α、P-tau 水平比較Note. Compared with blank group,*P<0.05. Compared with Aβ induction group,#P<0.05. Compared with miR-202-5p mimic group,△P<0.05.Figure 7 Comparison of PCSK9, TC, TNF-α, P-tau levels in cell supernatant
2.9 共轉染miR-202-5p 及pcDNA-PCSK9 對AD模型皮質神經元細胞miR-202-5p/PCSK9 軸及下游蛋白表達影響
與空白組相比,Aβ 誘導組細胞上清液中miR-202-5p、LRP-1、ApoE 表達降低,PCSK9、BACE1 蛋白表達升高(P<0.05)。 與Aβ 誘導組相比,miR-202-5p mimic 組、miR-202-5p mimic+pcDNA-NC 組、miR-202-5p mimic+pcDNA-PCSK9 組細胞miR-202-5p、LRP-1、ApoE 表達升高,PCSK9、BACE1 蛋白表達降低(P<0.05)。 與miR-202-5p mimic 組相比,miR-202-5p mimic+pcDNA-PCSK9 組LRP-1、ApoE 表達降低,PCSK9、BACE1 蛋白表達升高(P<0.05),miR-202-5p 表達差異不顯著(P>0.05)。 mimic-NC 組與模型組相比,miR-202-5p mimic 組與miR-202-5p mimic+pcDNA-NC 組相比,上述指標差異不顯著(P>0.05),見圖8。

圖8 腦皮質神經元細胞蛋白表達比較Note. A, Blank group. B, Aβ induced group. C, miR-202-5p mimic group. D, miR-202-5p mimic+pcDNA-NC group. E, miR-202-5p mimic+pcDNA-PCSK9 group. F, mimic-NC group. Compared with blank group,*P<0.05. Compared with Aβ induction group,#P<0.05. Compared with miR-202-5p mimic group,△P<0.05.Figure 8 Comparison of protein expression in cerebral cortex neurons
3 討論
隨著社會老齡化的加重,AD 已成為急需解決的世界難題。 據美國聯邦調查局統計,美國10 年間由于AD 導致的死亡人數已增加了146.2%;到本世紀中葉,美國65 歲以上的AD 病人可能增加至1380萬,而中國AD 病人將突破2800 萬,成為世界上AD病人最多的國家[12]。 AD 帶來的不僅是巨大的經濟負擔,還有死亡的威脅[13]。 故探尋有效的措施防止AD 發生、減緩AD 發展進程顯得尤為重要。
國際工作組已將腦脊液中Aβ 水平納入AD 診斷指標中[14]。 探究Aβ 生成及清除機制,可能對逆轉AD 患者Aβ 積聚引起的神經纖維纏結、神經元炎性損傷及丟失等神經系統損傷有重大研究價值[15]。大量文獻研究發現,AD 發生時,大量生成的BACE1可裂解并切割APP 的671 至672 間肽鍵,并在γ 分泌酶水解條件下生成Aβ40、Aβ42 而發揮神經毒性作用[16]。 本研究經腦內注射Aβ1-42 發現大鼠腦脊液中Aβ42、Aβ40 水平、腦皮層組織P-tau、炎癥因子TNF-α、IL-1β 水平均升高的同時,促Aβ42、Aβ40 生成的BACE1 表達也顯著升高,大鼠表現出神經元損傷、丟失及空間記憶及學習能力降低,提示造模成功。 Aβ1-42 體外誘導的神經元凋亡過程中,也檢測到BACE1 水平的升高,證實BACE1 切割APP 生成的Aβ,可能是AD 發生的關鍵因素。
近來研究發現,腦內膽固醇變化會影響Aβ 代謝并導致AD 神經變性[17]。 文獻研究報道發現,膽固醇是腦內髓鞘的重要組成部分,可參與神經元及突觸發育、神經炎性產生、受損細胞膜的維持和修復過程[18]。 但膽固醇不能透過血腦屏障而只能在大腦原位的局部神經系統合成[19],而隨著年齡的增長,成年神經元會逐漸失去合成膽固醇的能力,而不得不依賴星形膠質細胞產生膽固醇、并將膽固醇轉運至神經元[20]。 星形膠質細胞將膽固醇轉運至神經元的過程中,必須經過ApoE、LRP1 的顆粒轉運[21-22]。 但AD 患者腦脊液中大量產生的PCSK9可降解ApoE、LRP1,使神經元對膽固醇的攝取吸收減少,而神經元膽固醇的消耗增加、攝取吸收減少,勢必會導致Aβ 肽代謝及tau 蛋白磷酸化異常而引起神經元凋亡[23-24]。 Zhang 等[25]研究發現星形膠質細胞產生的膽固醇,不能被神經元攝取后,可在臨近脂筏、膜微域中積累,而為不溶性Aβ 片段提供穩定的沉積環境,并進一步誘導Aβ 的產生,認為膽固醇穩態變化,是影響Aβ 產生的關鍵因素,這與楊宏艷等[26]膽固醇轉運抑制劑加重星形膠質瘤細胞促BACE1-APP-Aβ 生成及AD 發展,觀點相一致。PCSK9 是參與膽固醇調節的重要因子[27],近來研究發現AD 患者PCSK9 的大量產生,可能與BACE1 切割APP 介導Aβ 生成活性升高有關,認為BACE1 大量升高可能刺激PCSK9 的大量產生,來抑制BACE1切割APP 并阻斷Aβ 生成來發揮神經保護作用[28]。但也有研究發現,PCSK9 的產生不僅可誘導Bcl/Bax-Caspase9 凋亡活性的升高來促進神經凋亡,還可降解并抑制ApoE、LRP1(具有促Aβ 清除、促神經元膽固醇攝取)表達,來加速Aβ 生成并阻礙其清除,而參與AD 發生發展過程[29-30]。 Xue 等[31]及Abuelezz 等[6]等發現,AD 患者PCSK9 的高度表達,在降低LRP-1 引起神經元膽固醇攝取減少及Aβ 清除受損的同時,還誘導TOLL 樣受體4 通路活化而導致炎癥因子TNF-α、IL-1β 分泌,進而導致神經元炎性損傷。 本研究也在AD 大鼠腦脊液及體外AD模型細胞上清液中,檢測到PCSK9 表達的升高,皮層組織及神經細胞中ApoE、LRP1 表達的降低,用PCSK9 抑制劑-Aliro 可明顯抑制PCSK9 升高、ApoE及LRP1 降低,而降低Aβ 生成、發揮抗AD 神經元炎性損傷癥狀,提示干預PCSK9 表達,可緩解AD過程中Aβ 沉積及神經損傷。
miRNA 也是參與AD 患者Aβ 生成、清除及神經元損傷的關鍵調節因子。 大量文獻研究發現,多種miRNA 可通過抑制BACE1 或APP 的產生,而抑制Aβ 沉積及AD 神經損傷[32]。 miRNA 中的miR-202-5p 可作為抑癌基因而參與乳腺癌、前列腺癌、口腔癌等腫瘤細胞異常增殖過程,近來研究發現,在腦缺血及神經元應激性凋亡過程中,miR-202-5p表達也異常降低,并認為miR-202-5p 異常表達也參與神經系統損傷過程[33]。 Dong 等[8]發現miR-202-5p 在AD 患者血清中表達降低,且miR-202-5p 與APP 的3’UTR 端有靶向結合位點,并認為miR-202-5p 發揮抗AD 作用,可能與靶向抑制APP 表達而阻斷Aβ 生成有關。 但miRNA 可通過調節多個靶蛋白發揮生物學效應,miR-202-5p 的抗AD 作用,除APP 外還可能通過其他靶蛋白發揮作用。 本研究發現,miR-202-5p 也與PCSK9 之間存在靶向結合位點,過表達miR-202-5p 可通過抑制PCSK9 表達來上調ApoE、LRP1 表達,發揮抗胞外膽固醇分泌、抗Aβ 生成及AD 神經炎性損傷作用。 上調PCSK9 表達后,可部分減弱miR-202-5p 的抗Aβ 生成及神經炎性因子分泌作用。
綜上所述,過表達miR-202-5p 可通過抑制PCSK9 活化抑制Aβ 生成并促進LRP-1/ApoE 介導的促膽固醇攝取及Aβ 清除,進而發揮抗AD 神經損傷作用。 這為AD 的基因靶向治療提供一定參考。但PCSK9 與BACE1-Aβ 生成途徑的調控關系還存在爭議較多,ApoE-LRP1 促膽固醇攝取及Aβ 清除作用盲點也較多,這需進一步深入研究。
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
網絡安全與數據管理(2022年1期)2022-08-29 03:15:20
導航定位學報(2022年4期)2022-08-15 08:27:00
中學生數理化·中考版(2022年8期)2022-06-14 06:55:24
新世紀智能(數學備考)(2021年9期)2021-11-24 01:14:36
成都醫學院學報(2021年2期)2021-07-19 08:35:14
新世紀智能(數學備考)(2020年9期)2021-01-04 00:25:14
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
數學物理學報(2020年2期)2020-06-02 11:29:24
光學精密工程(2016年6期)2016-11-07 09:07:19
