藿香正氣丸(濃縮丸)HPLC 指紋圖譜
2022-12-25 04:53:42王孟穎孫福英趙雯饒毅萬林春
藥品評價 2022年19期
王孟穎,孫福英,趙雯,饒毅,萬林春
1.江西省藥品檢驗檢測研究院,國家藥品監督管理局中成藥質量評價重點實驗室,江西省藥品與醫療器械質量工程技術研究中心,江西 南昌 330029;2.江西省腫瘤醫院,江西 南昌 330029;3.江西中醫藥大學,江西 南昌 330004
[關鍵字]藿香正氣丸(濃縮丸);指紋圖譜;相似度評價;HPLC
藿香正氣丸(濃縮丸)是一種解表化濕藥,收載于《中華人民共和國衛生部藥品標準中藥成方制劑》第7 冊[1],其中僅收載了性狀、顯微鑒別等項目,尚無含量限定的相關項目。為更好地控制藥品質量,許多研究者對藿香正氣系列進行了研究,黃淡霞[2]建立起了藿香正氣丸中的厚樸、廣藿香等六味藥材的TLC 鑒別方法和橙皮苷、厚樸酚的HPLC 含量測定方法。林雀躍等[3]為藿香正氣合劑建立了專屬的HPLC指紋圖譜,最終確定了15個共有峰。陳宗良等[4]使用超高效液相色譜建立了藿香正氣水的指紋圖譜,并標定了9 個特征峰。但由于各種藿香正氣丸的劑型和配方略有不同,因此亟待建立藿香正氣丸專屬的指紋圖譜。本研究組選取4 個具有代表性的生產廠家的16 批藿香正氣丸(濃縮丸)進行測定后,建立了HPLC 指紋圖譜,并對30 批藿香正氣丸(濃縮丸)進行指紋圖譜的測定并計算相似度。結果表明相似度良好,能有效控制藿香正氣丸(濃縮丸)質量。
1 儀器與試劑
1.1 儀器
Agilent 1260 系列(全自動)高效液相色譜儀,Chemstation 化學工作站,DAD 檢測器;Sartorius BSA 124S-CW 電子天平;Sartorius BT 25S 電子天平。
1.2 試劑
甘草苷、異歐前胡素、橙皮苷、甘草酸銨、歐前胡素、和厚樸酚、厚樸酚均購于中國食品藥品檢定研究院,批號分別為110610-200604、110827-200407、110721-201616、110731-201418、110826-200712、110730-201614、110729-201513。甘草素、川陳皮素購于上海安譜實驗科技股份有限公司,批號分別為T1670010、T3560010。乙腈為色譜純級,水為超純水,其余試劑為分析純。
2 方法與結果
2.1 色譜條件
Agilent Zorbax C18(250 mm×4.6 mm,5 μm);流動相:乙腈(A)-0.2%甲酸溶液(B),按表1程序梯度洗脫;檢測波長270 nm;柱溫為35℃;流速為 1.0 mL/min。理論板數按橙皮苷峰計算應不低于5 000。

表1 流動相梯度洗脫表
2.2 溶液制備
2.2.1 供試品溶液的制備 取藿香正氣丸(濃縮丸),于研缽中研細,取粉末約2 g,置具塞錐形瓶中,精密稱定,精密加入甲醇25 mL,稱定重量,超聲處理(功率:500 W,頻率:40 kHz)30 min,放冷,再稱重,用甲醇補足減失的重量,用微孔濾膜(0.45 μm)濾過,取續濾液,即得。
2.2.2 對照品溶液的制備 取對照品適量,精密稱定,加甲醇制成每1 mL 各含20 μg 的混合溶液,即得。
2.3 測定方法
分別精密吸取對照品溶液與供試品溶液各10 μL,進樣測定,記錄色譜圖。以3 號色譜峰(橙皮苷)作為參照。供試品指紋圖譜中應呈現與對照品保留時間相同的色譜峰。按中藥指紋圖譜相似度評價系統計算,供試品與對照指紋圖譜的相似度應大于等于0.85。
2.4 方法學考察
2.4.1 精密度實驗 取藿香正氣丸(濃縮丸)(批號:20150614)1 份,按“2.2.1”項下方法制備供試品溶液,連續進樣6 次。以3 號共有峰(橙皮苷)作為參照峰,計算各色譜峰保留時間的RSD 為0.02%~1.67%,相對峰面積的RSD 為0.02%~1.86%。
2.4.2 重復性實驗 取藿香正氣丸(濃縮丸)(批號:20150614)6 份,按“2.2.1”項下方法制備供試品溶液,進樣測定。以3 號共有峰(橙皮苷)作為參照峰,計算各色譜峰保留時間的RSD 為0.04%~1.72%,相對峰面積的RSD 為0.02%~1.92%。
2.4.3 穩定性實驗 取藿香正氣丸(濃縮丸)(批號:20150614)1 份,按“2.2.1”項下方法制備供試品溶液,分別于0 h、2 h、4 h、8 h、12 h 進樣測定。以3 號共有峰(橙皮苷)作為參照峰,計算各色譜峰保留時間的RSD 為0.02%~0.75%,相對峰面積的RSD 為0.02%~1.91%。
2.5 指紋圖譜的建立及相似度評價
選取具有代表性的4 個生產廠家的樣品(共16批)建立共有模式,按“2.2.1”項下方法制備供試品溶液,“2.1”項下色譜條件進樣測定,圖譜導入2012 版《中藥色譜指紋圖譜相似度評價系統》,選取“時間窗寬度”為0.10 min,以“中位數”生成對照圖譜,自動校準后,得到藿香正氣丸(濃縮丸)的標準對照圖譜。見圖1。經與對照藥材和對照品的色譜峰對比,最終共標定了13 個共有峰,并指認出9個成分,對照品色譜峰見圖2~10。參考《中國藥典》血脂康膠囊等[5]品種項下指紋圖譜的限度與相關文獻[6-10],同時考慮到不同產地原藥材成分含量的差異以及不同企業工藝控制水平的差異,暫擬定供試品指紋圖譜與對照指紋圖譜的相似度限度為0.85。
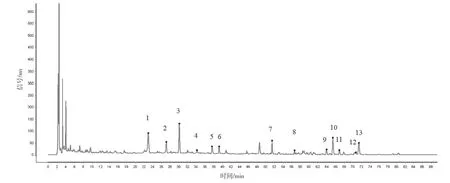
圖1 藿香正氣丸(濃縮丸)HPLC指紋圖譜
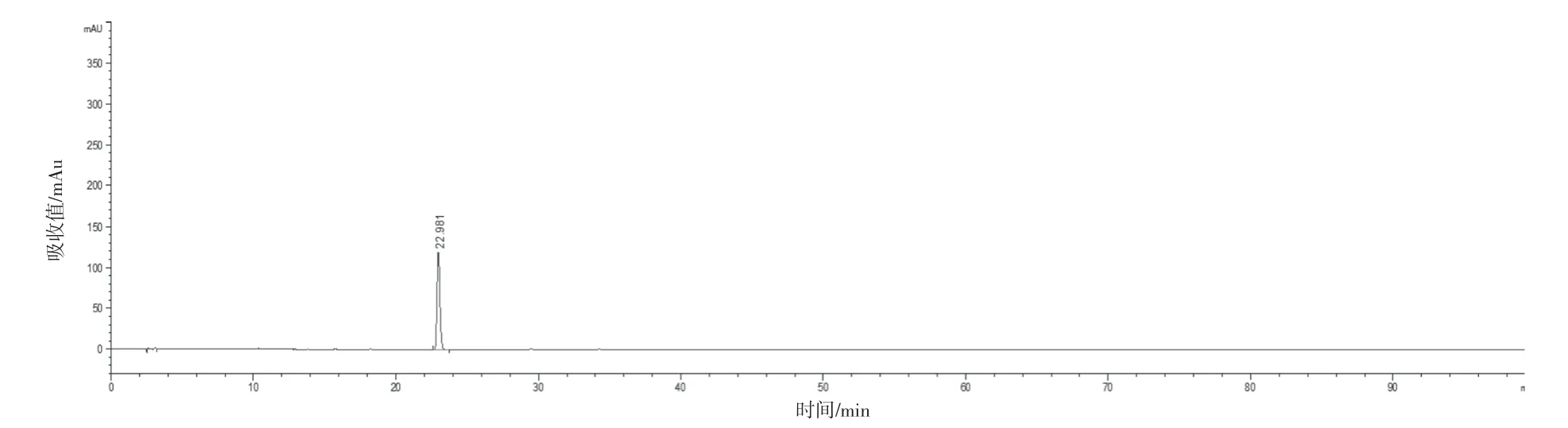
圖2 甘草苷對照品色譜圖

圖3 橙皮苷對照品色譜圖
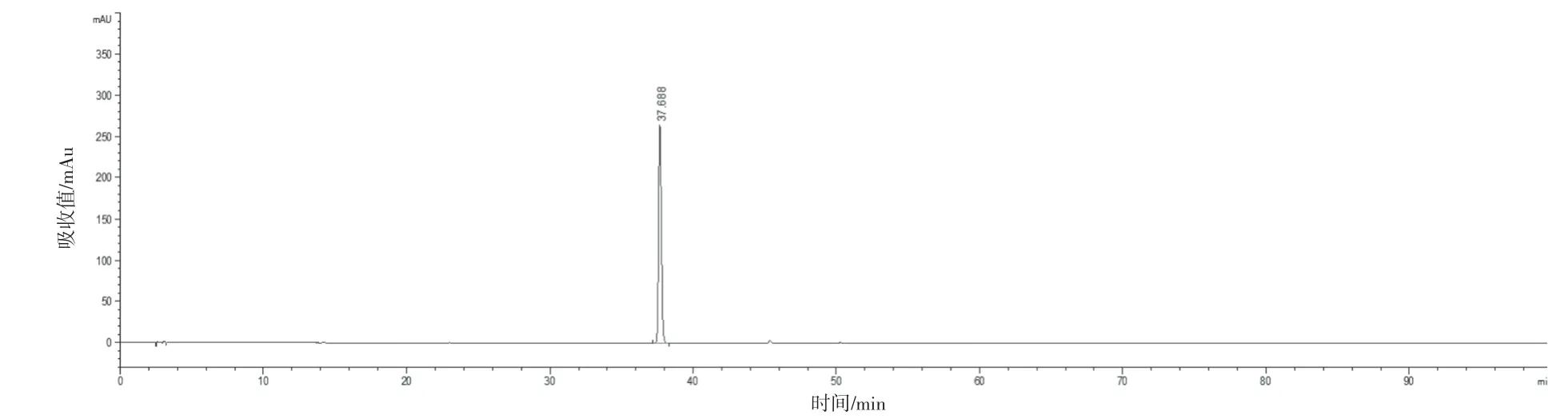
圖4 甘草素對照品色譜圖

圖5 甘草酸銨對照品色譜圖
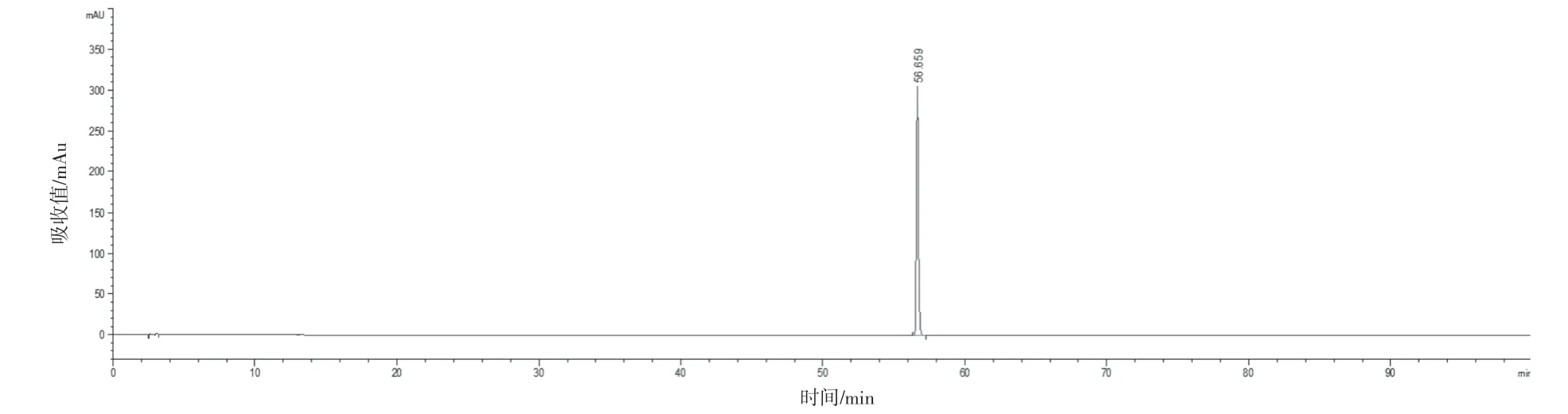
圖6 川陳皮素對照品色譜圖

圖7 歐前胡素對照品色譜圖

圖8 和厚樸酚對照品色譜圖

圖9 異歐前胡素對照品色譜圖

圖10 厚樸酚對照品色譜圖
運用該藿香正氣丸(濃縮丸)HPLC 指紋圖譜方法,測定了9 個企業的30 批藿香正氣丸(濃縮丸),結果顯示這30 批樣品的指紋圖譜與對照指紋圖譜的相似度均高于0.85,結果見表2。

表2 藿香正氣丸(濃縮丸)生產廠家、批號及相似度
3 討論
3.1 檢測波長的選擇
由于不同的化學結構,不同化學物質的紫外吸收波長各有不同。本研究中確定的9 種化合物的最大吸收波長在254~336 nm 范圍內,集中分布于
254~294 nm 波長范圍內,為使本實驗各峰的響應值都較大,最終確定檢測波長為270 nm。
3.2 提取溶劑和提取方法的考察
本研究考察了甲醇、甲醇-鹽酸(100∶1)兩種提取溶劑和超聲30 min、回流1 h 兩種提取方法對實驗結果的影響。結果顯示,不同溶劑、不同提取方式的色譜結果中,各個峰的響應值相當。為使操作更簡便,最終確定以甲醇為溶劑,超聲提取30 min。
本研究建立了藿香正氣丸(濃縮丸)的HPLC指紋圖譜,最終共標定了13 個共有峰,并指認出9 個成分,通過2012 版中藥色譜指紋圖譜相似度評價系統計算相似度,結果表明不同廠家間的藥品存在一定差異,這可能是不同的生產工藝、原料來源造成的。通過方法學驗證,HPLC 指紋圖譜的方法結果穩定、可靠,可以用于藿香正氣丸(濃縮丸)的質量控制,HPLC 指紋圖譜的建立也為藿香正氣丸(濃縮丸)的產品一致性評價提供了參考。
