柱前衍生-高效液相色譜法測定嬰幼兒食品和乳品中膽堿含量
2023-01-13 12:39:22盧越圻金志浩袁雯菲
工業微生物 2022年6期
關鍵詞:檢測
盧越圻, 孫 潔*, 金志浩, 薛 麟, 袁雯菲
1.中檢科(上海)測試技術有限公司,上海 201206;2.通標標準技術服務(上海)有限公司,上海 200233
膽堿具有促進大腦發育、促進神經系統的信息傳遞等功能[1]。膽堿通常是以氯化膽堿或酒石酸膽堿的形式添加到嬰幼兒配方食品中的。我國食品安全國家標準GB 10765—2010[2]、GB 10767—2010[3]等對嬰幼兒配方食品和其他食品中膽堿的含量有明確的限量規定。目前對食品中膽堿含量的檢測主要包括雷氏鹽比色法[4]、酶比色法[4-6,11]、離子色譜法[11]、HPLC-MS法[7,11]、HPLC-ELSD法[8]、UPLC-FLD法[9,10]等。其中酶比色法(如國標GB 5413.20—2022第一法)使用較多、較為經典,但是成本高、效率低、背景干擾因素較多,且不適用于試樣中維生素C含量大于100 mg/100 g的樣品[6],現有的國標和文獻里暫時未出現使用本方法用于膽堿的測定,因此本文首次嘗試使用高效液相色譜法測定膽堿含量,彌補國標方法檢測膽堿含量成本高、耗時長、工作量大、維生素C等還原性成分過高影響檢測等方面的不足。
1 材料與方法
1.1 儀器與試劑
膽堿酒石酸氫鹽標準品(Dr.)、芴甲氧羰酰氯(Fmoc-Cl)、乙腈(色譜純)、硼酸、氫氧化鈉、磷酸(優級純)、正己烷(色譜純)、十二水合磷酸氫二鈉(色譜純)、十水合四硼酸鈉(色譜純)、鹽酸(優級純)(除注明外均為分析純)。衍生劑:10 mg/mL的Fmoc-Cl乙腈溶液。緩沖液:0.4 mol/L的硼酸鈉緩沖液(pH 11.0)。流動相A:十二水合磷酸氫二鈉9.0 g,十水合四硼酸鈉9.5 g,加水2 L,用鹽酸調pH至8.2。流動相B:乙腈。
賽默飛液相色譜儀U3000配熒光檢測器、日本島津RF-6000熒光分光光度計。
1.2 色譜條件
色譜柱:Agilent Eclipse XDB-C18 Column(4.6 mm×250 mm,5 μm);流動相:流動相A∶流動相B=65∶35;流速:1.0 mL/min;檢測波長:熒光波長:激發波長265 nm,發射波長315 nm;柱溫:40 ℃;進樣量:4 μL(程序進樣)。
1.3 標準曲線的制作
膽堿(以膽堿氫氧化物計)標準儲備溶液(2 500 mg/L):稱取在102 ℃±2 ℃烘至恒重的膽堿酒石酸氫鹽522.5 mg,用水溶解并轉移至100 mL容量瓶中定容,混勻。繼續用水逐級稀釋成1 μg/mL、2 μg/mL、4 μg/mL、6 μg/mL、8 μg/mL、10 μg/mL、20 μg/mL的標準曲線。
1.4 樣品的測定
稱取5 g~10 g混合均勻的試樣,于100 mL的錐形瓶中,加入30 mL 1 mol/L鹽酸溶液。將裝有試樣的容器放在70 ℃水浴中,加塞混勻,水解3 h(每隔30 min振搖一次),冷卻。用氫氧化鈉溶液調pH為5.5~6.0,轉入50 mL容量瓶中,用蒸餾水定容至刻度,過濾水解液,膽堿濃度太大時稀釋至標曲范圍內。同時做空白試驗。
2 結果與討論
2.1 衍生條件的優化
2.1.1衍生終止劑和萃取劑
用正己烷萃取水解產物等雜質,樣液比為試樣溶液∶緩沖液∶衍生試劑∶衍生終止劑(0.1%的磷酸溶液)∶正己烷=400 μL∶200 μL∶200 μL∶200 μL∶2 mL(×3次),在室溫下衍生,衍生時間為5 min。使用1.2液相條件,進樣量10 μL。試驗結果顯示:1)使用衍生終止劑對結果的影響較大,會使結果偏小;2)使用正己烷萃取過量的Fmoc-Cl及其水解產物FMOC-OH(隨著衍生時間的增加反應會趨向于Fmoc-Cl的水解,其產物為FMOC-OH)的同時會萃取一定量的衍生物,導致結偏小。因此確定,衍生的比例為試樣溶液∶緩沖液∶衍生試劑=2∶1∶1,不使用衍生終止劑和正己烷。
2.1.2正交試驗分析
在此基礎上,以20 μg/mL的工作液色譜峰面積為評價指標,設計L18(37)正交試驗,對硼酸緩沖液pH(A)、衍生劑濃度c(B)、衍生溫度T(C)和衍生時間t(D)4個影響因素進行考察,每個因素選取三個水平,考察各因素的主次影響順序和最優水平組合。實驗操作步驟同2.1.1。正交試驗因素見表1、正交試驗數據分析結果見表2。
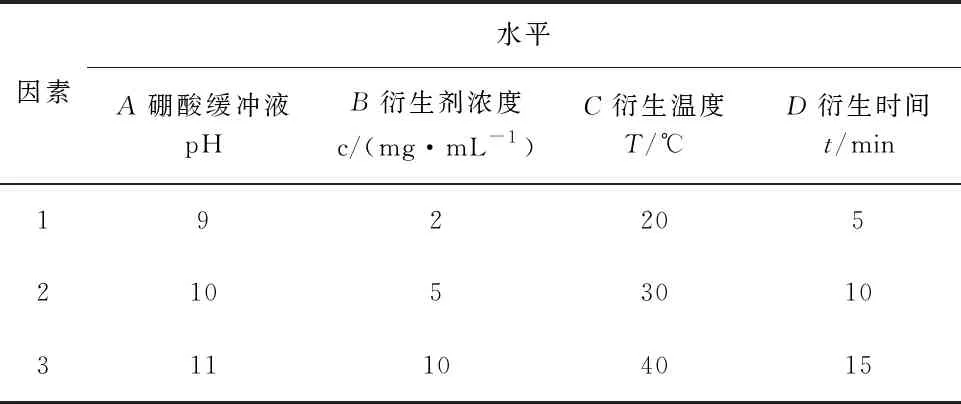
表1 膽堿衍生化條件因素水平表
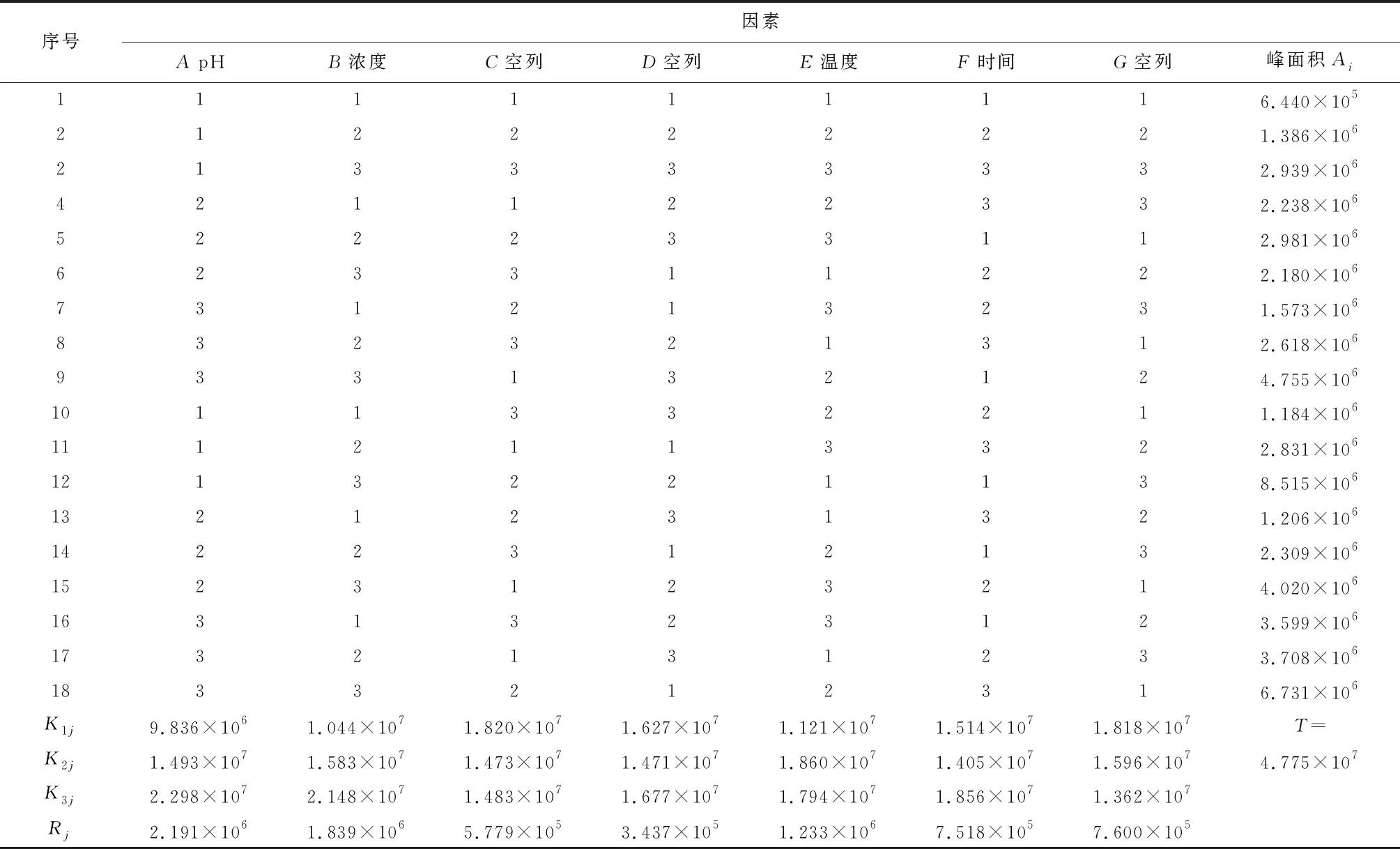
表2 膽堿衍生化條件正交試驗數據分析結果
通過極差分析,各因素的主次順序為A>B>C>D,可初步得出的最優方案為A3、B3、C2、D3。進一步的方差分析顯示四個因素F值均不顯著(p>0.05),這可能是由于誤差自由度過小,分析的靈敏度不夠高的緣故。對主要因素A、B用LSR法進行多重比較,得到以A3、B3為最好。綜合極差分析和方差分析結果,本試驗的最優處理組合為A3、B3、C2或C3、D3或D1,本文選用A3、B3、C2、D3,即硼酸緩沖液pH為11,衍生劑濃度10 mg/mL,衍生溫度30 ℃,衍生時間15 min。
2.2 色譜條件的優化
實驗操作步驟同2.1.1,優化色譜條件。
2.2.1檢測波長的確定
使用紫外檢測器對衍生物進行全波長掃描,綜合考慮靈敏度、基線波動、其他雜質組分被檢測出的可能性,最終選擇265 nm為紫外檢測波長。在此基礎上,以265 nm為激發波長,利用熒光分光光度計掃描確定最佳激發波長。通過掃描發現在315 nm處響應最大,故確定發射波長為315 nm。因此,熒光檢測波長選用激發波長為265 nm,發射波長為315 nm。由于在試驗中發現選用紫外檢測器測定膽堿衍生物時響應很小,不利于定量,因此在實際測定時選用熒光檢測器檢測。
2.2.2流動相的選擇
考察不同流動相比例下的色譜峰分離情況。結果顯示,在流動相A比例大于70%的情況下,未檢測到衍生物組分,說明衍生物組分不能被洗脫;而流動相A比例小于50%的情況下,衍生物組分的保留時間小于5 min,因此綜合考慮實際樣品中各組分分離度和檢測時間,確定選擇流動相為A∶B=65∶35。
2.3 方法學驗證
2.3.1衍生產物的專屬性
分別吸取空白溶液、20 μg/mL膽堿工作液、一奶粉樣品水解處理溶液各400 μL,實驗操作步驟同2.1.1。結果表明,試樣中膽堿經衍生化反應后其色譜峰理論塔板數能夠達到14 000,分離度能夠達到2.3,拖尾因子能夠達到1.1,空白在保留時間處無干擾,說明C18色譜柱對其有較好的分離效能,膽堿衍生物與其他被分離物質的分離程度好,膽堿衍生物的色譜峰略有拖尾,但對試驗影響不大。綜上所述,該方法專屬性較強,可以用來測定膽堿的含量。
2.3.2衍生產物的穩定性
吸取20 μg/mL膽堿工作液400 μL,實驗操作步驟同2.1.1,每20 min測定一次,考察在室溫下衍生溶液的穩定性。結果表明,衍生物性質不穩定,需要在衍生結束后立即測定。因此為了保證衍生物的穩定測定以及批量測定,研究使用HPLC的程序進樣功能實現柱前自動衍生。結合之前的試驗結果和儀器特性,對UdpMixWait程序的設置做了考察,UdpMixWait是4 min~30 min,按照試樣溶液∶緩沖液∶衍生試劑=2 μL∶1 μL∶1 μL進行衍生,使用1.2液相條件。結果顯示,UdpMixWait在20 min之后衍生物峰面積開始逐漸變小,說明衍生物開始分解,結合2.1所確認的衍生條件以及實際樣品測定的可行性,最終確定15 min較為合適。
利用以上確定的程序進樣條件對20 μg/mL的膽堿工作液進行測定,參考JJG 705—2014[12]的相關要求,考察其定性、定量重復性。結果表明,衍生產物的定性重復性為0.15%、定量重復性為2.43%,達到了預期效果,利用程序進樣對目標物進行衍生的操作是可行的,保證了衍生物的穩定測定以及批量測定的可能性。
2.4 線性關系、最低檢出限、定量限
標準曲線:對1.3的標準曲線進行測定,以濃度為橫坐標,以峰面積為縱坐標,繪制標準曲線。結果顯示,膽堿濃度在1 μg/mL~20 μg/mL的范圍內有較好的線性關系,回歸方程為y=311 343.549 7x+347 196.959 4,相關系數R2=0.999 7。20 μg/mL的膽堿衍生物的色譜峰見圖1。

圖1 膽堿衍生物色譜圖
最低檢出限、定量限:結果顯示0.3 μg/mL及1 μg/mL的膽堿工作液的S/N分別為7.255及17.117,能達到3 S/N及10 S/N,按照稱樣量為5 g,定容體積為50 mL,計算出方法的LOD為0.3 mg/100 g、LOQ為1 mg/100 g。
2.5 回收率及精密度實驗
在2.1、2.2、2.3實驗條件下,對4個不同樣品分別添加低、中、高三個濃度水平的標樣,進行三水平六平行加標回收實驗,計算平均值、測定回收率及精密度,其中奶粉2維生素C含量大于100 mg/100 g。結果顯示,各樣品的平均加標回收率為 85.0%~110.4%,相對標準偏差(RSD)為1.27%~3.78%,結果見表3。
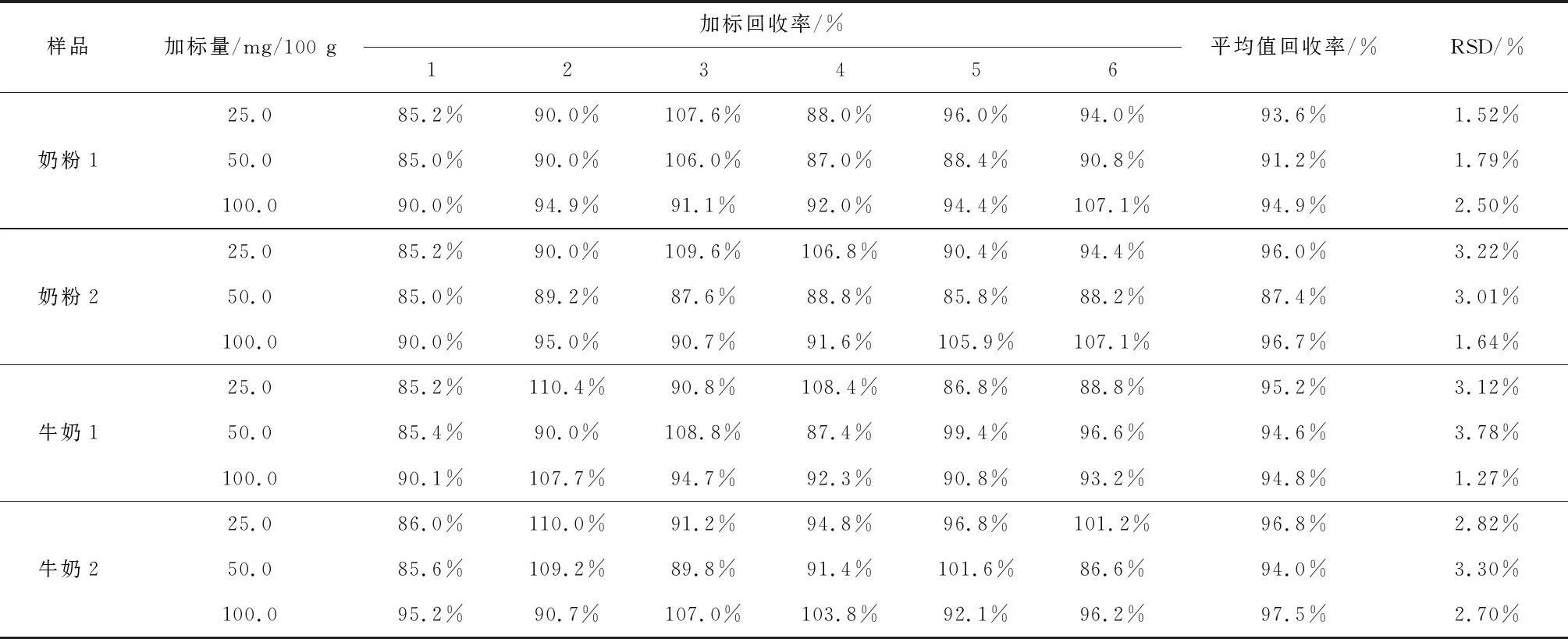
表3 方法回收率、精密度測定結果(n=6)
2.6 本方法與國標的比較
用國標GB 5413.20—2022(第一法 酶比色法)與本方法對3個不同樣品分別進行測定,并且分別對樣品進行兩向分組有相等重復觀測值試驗資料的方差分析,以國標測定數據為基準,比較兩個檢測方法的數據差異,結果見表4及表5。結果表明,本法和國標相比,同一樣品不同方法數據間差異不顯著(p>0.05),本法的測定數據是可以接受的,說明本法能較為有效的測定樣品中膽堿的含量。
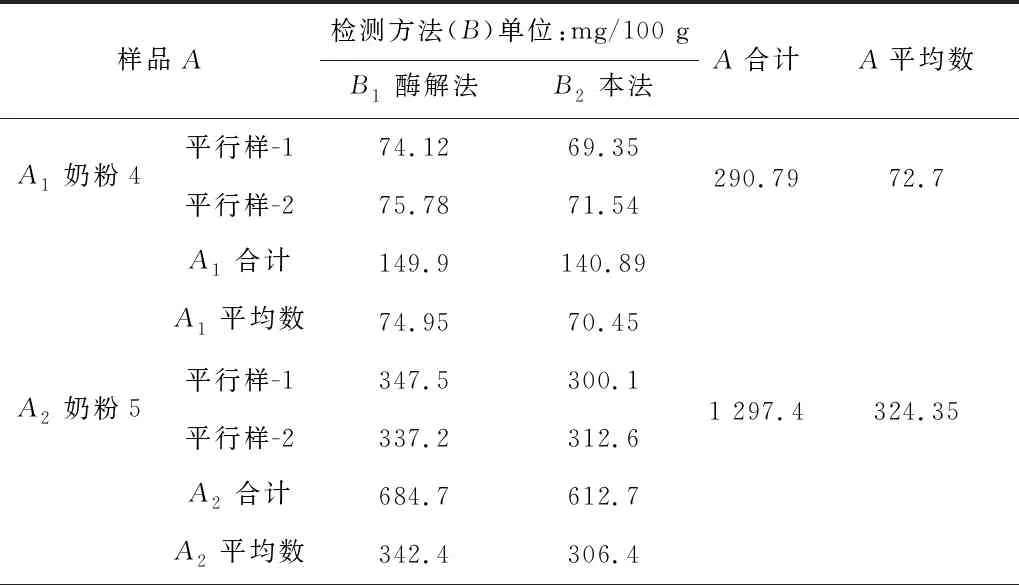
表4 本法和國標測定樣品數據對比
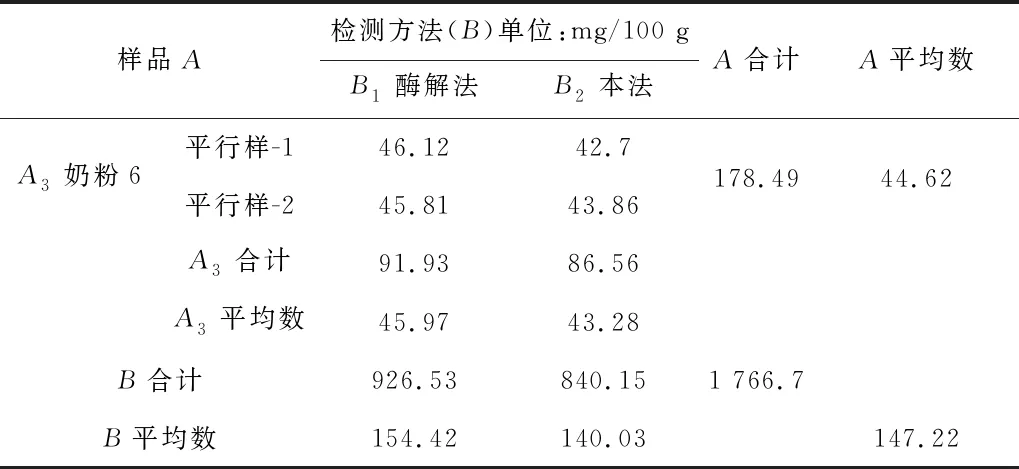
表4(續)
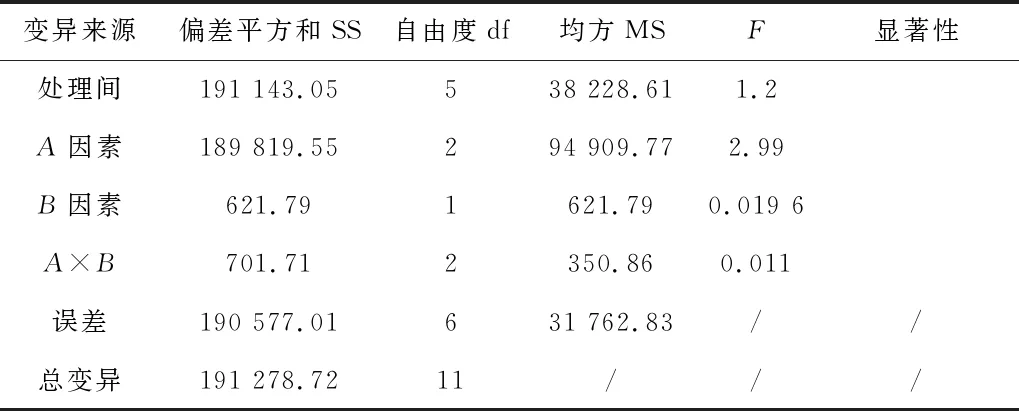
表5 本法和國標測定樣品數據方差分析表
3 結論
本文通過正交試驗得到利用Fmoc-Cl衍生膽堿的最佳條件,利用高效液相色譜儀的程序進樣功能實現在線衍生,首次建立了使用高效液相色譜-熒光檢測器測定膽堿含量的方法。利用該方法建立的標準曲線線性良好,LOD為0.3 mg/100 g、LOQ為1 mg/100 g。樣品平均加標回收率為85.0%~110.4%,RSD為1.27%~3.78%。相比于傳統的酶解法,本法在試劑成本上可以降低90%,在樣液制備完成后省去了酶解、紫外上機等人工操作的時間;目標物與其他雜質可被C18柱有效分離。通過比較本方法和國標GB 5413.20—2022第一法對相同樣品進行測定的數據,結果顯示兩種方法的數據間差異不顯著(p>0.05),本法能有效對樣品進行膽堿的測定。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
