細菌感染激活炎癥小體介導細胞焦亡的機制研究進展
2023-01-28 03:47:44李世青袁媛王景林
中國免疫學雜志 2022年21期
李世青 袁媛 王景林
(軍事醫學研究院微生物流行病研究所,病原微生物生物安全國家重點實驗室,北京 100071)
炎癥小體作為一種胞質大分子信號平臺,自2002年提出以來逐漸走入研究者視野,作為機體免疫系統的重要組成備受關注。宿主細胞受到細菌感染后,細菌不同組件可通過不同機制激活宿主體內多種炎癥小體組裝,引發炎癥反應。炎癥反應是把雙刃劍,在感染早期,炎癥反應有利于機體抵御細菌感染,但持續性的過度炎癥反應則有害于機體,導致多器官衰竭。本文綜述了不同細菌組件激活炎癥小體的種類并同時闡述了各炎癥小體的激活機制。
1 炎癥小體介導細胞焦亡的概述
機體受到外來刺激時,宿主細胞可識別特定刺激物,引發體內炎癥小體組裝及其介導的炎癥反應。炎癥小體介導的炎癥反應主要表現為兩方面:一方面組裝好的炎癥小體能夠調節caspase-1活化,在天然免疫防御過程中促進細胞因子前體pro-IL-1β和pro-IL-18成熟和分泌。另一方面,炎癥小體組裝復合物能夠調節caspase-1依賴的炎癥形式編程性細胞死亡——細胞焦亡(pyroptosis),誘導細胞在炎癥和應激病理條件下死亡。炎癥小體介導的兩方面炎癥反應既有區別,也存在聯系。本文將分別介紹炎癥小體及細胞焦亡,并進一步闡述兩者的聯系。
1.1 炎癥小體炎癥小體于2002年由TSCHOPP實驗組提出,從結構來說,其是由傳感器、適配器ASC(apoptosis-associated speck-like protein containing a CARD)和酶原pro-caspase-1組成的多聚蛋白復合物[1]。傳感器包括三類蛋白,分別為NLR[nucleo?tide-binding oligomerization domain(NOD)-like recep?tor]家族成員、AIM2(absent in melanoma 2)蛋白和Pyrin蛋白。以傳感器為分類依據,目前研究最為廣泛的炎癥小體主要包括NLRP1、NLRP3、NLRC4、AIM2、Pyrin。NLRs由三部分組成:C末端、中央高度保守的可結合核苷酸的寡聚結構域(nucleotidebinding domain,NACHT)及可結合下游蛋白的可變N端。C末端為LRR(leucine rich repeat)結構域,能夠識別細胞溶質配體并將NLRs保留在非激活狀態。依據不同N末端結構域,NLR家族蛋白可進行亞分類,如帶有PYD(Pyrin domain)的NLRP、帶有BIR結構域(baculovirus inhibitory repeat domain)的NLRB和帶有CARD(caspase activation and recruit?ment domain)的NLRC。AIM2屬于ALR(AIM2-like receptor)模式識別受體家族蛋白成員,其N端含有PYD結構域,C端含有負責結合多聚核苷酸的HIN200結構域。Pyrin蛋白主要由3個結構域組成:N端Pyrin域(PYD)、2個中心鋅指域(B-boxes)、1個螺旋線圈域(coiled-coil,CC)。適配器ASC蛋白由PYD-CARD組成,在炎癥小體組裝過程中起中間橋梁作用。識別特定刺激后,傳感器與ASC結合,在細胞核周圍形成1個ASC斑點結構,隨后促進procaspase-1聚集,進而引發體內炎癥小體組裝及其介導的炎癥反應。
1.2 細胞焦亡細胞焦亡是近年發現的一種不同于凋亡、壞死的細胞死亡方式,主要表現為細胞不斷腫脹直至細胞膜破裂,導致細胞內容物釋放進而激發強烈炎癥反應。目前認為細胞焦亡存在兩種途徑,一類是caspase-1參與組成的炎癥小體引發的經典細胞焦亡途徑,另一類是小鼠caspase-11和人caspase-4/5單獨作為胞質傳感器響應胞質脂多糖(LPS)引起的非經典細胞焦亡途徑。最初發現cas?pase-1/4/5/11可裂解GSDMD,釋放其N端結構域,在細胞膜上寡聚成孔,誘發細胞焦亡[2]。與上述不同的是,近年也有研究表明耶爾森氏菌(Yersinia)感染巨噬細胞后,caspase-8可水解活化Gasdermin引發細胞焦亡[3]。也有研究表明caspase-3可通過酶切GSDME激活細胞焦亡[4]。因此,目前細胞死亡命名委員會建議細胞焦亡定義為一種依賴于Gasdermin家族蛋白形成質膜膜孔的可調控細胞死亡,通常(但并不總是)是炎癥性caspase活化的結果[5]。
1.3 炎癥小體與細胞焦亡的聯系機體受到細菌感染后可識別細菌刺激物激活體內炎癥小體組裝引發細胞焦亡,介導炎癥反應發生。炎癥小體與細胞焦亡既存在聯系也存在差別。兩者聯系主要表現為兩方面:一是炎癥小體可作為經典細胞焦亡途徑的上游通路引發細胞焦亡;二是兩者分別對細胞因子IL-1β和IL-18的成熟和分泌發揮作用。刺激物激活炎癥小體可引起pro-IL-1β和pro-IL-18成熟,而其進一步釋放依賴于細胞焦亡引發的膜孔形成。兩者相互依存,共同促發體內炎癥反應。本綜述從細菌組件出發,介紹不同細菌組件可激活經典炎癥小體(NLRP1、NLRC4、NLRP3、AIM2、Pyrin)的種類,同時探討這些炎癥小體激活介導經典細胞焦亡的發生機制。
2 不同細菌組件可激活炎癥小體的種類
不同細菌感染可激活不同炎癥小體,同一細菌感染也可激活不同炎癥小體,體現細菌感染激活炎癥小體的多樣性與網絡性。宿主細胞感應不同細菌組件,激活不同炎癥小體,引發細胞焦亡和細胞因子釋放,具體細菌組件激活炎癥小體介導細胞焦亡的匯總見表1、表2。以下將以細菌菌體表面結構、附件、內部物質、分泌系統效應蛋白及外毒素為分類依據,匯總這些組件可激活炎癥小體的種類。
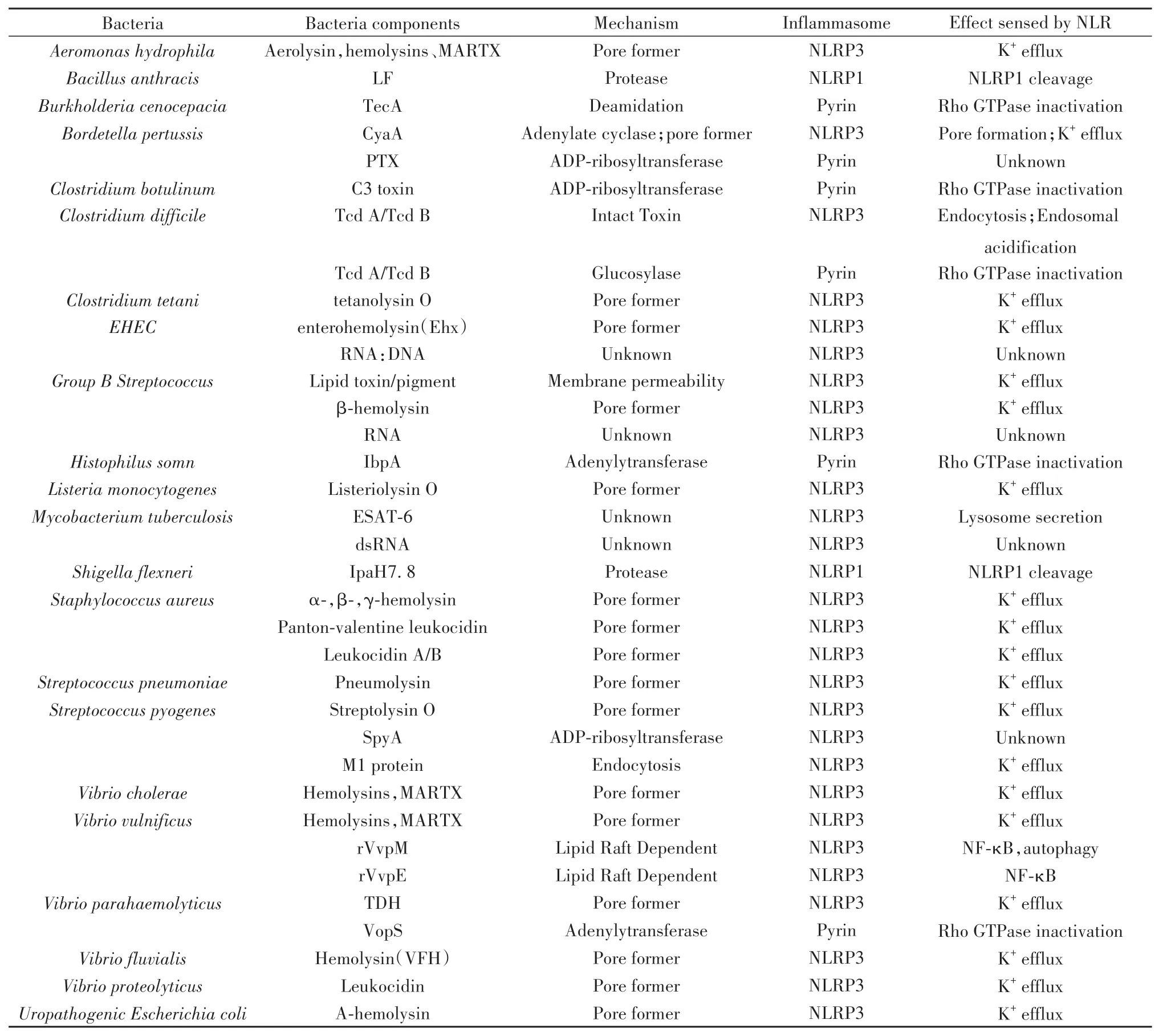
表1 細菌感染間接激活炎癥小體[6-7]Tab.1 Bacterial infection indirectly activates inflammasome[6-7]
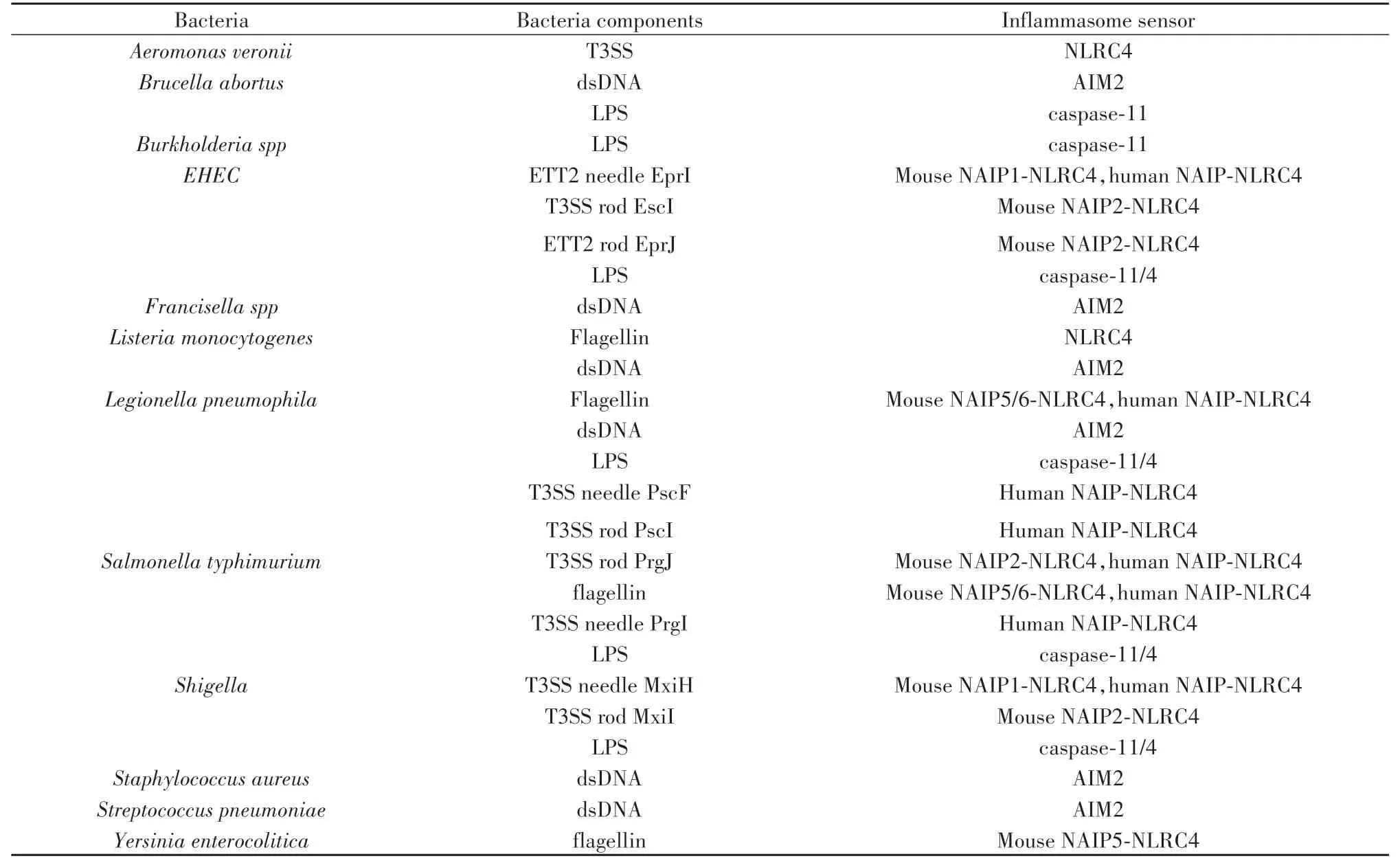
表2 細菌感染直接激活炎癥小體[7-8]Tab.2 Bacterial infection directly activates inflammasome[7-8]
2.1 菌體表面結構(細胞壁、表面蛋白)可激活炎癥小體的細菌表面結構主要包括細胞壁成分、表面蛋白。如干酪乳桿菌(Lactobacillus casei)細胞壁片段、化膿鏈球菌(Streptococcus pyogenes)細胞表面蛋白M1可激活NLRP3炎癥小體引發經典細胞焦亡途徑[9-10]。此外,革蘭氏陰性菌細胞壁成分LPS中的脂質A可直接與caspase4/5/11結合引發非經典細胞焦亡途徑[11]。
2.2 菌體附件(鞭毛)可激活炎癥小體的細菌附件是細菌的鞭毛結構,如軍團菌(Legionella)、沙門氏菌(Salmonella)、假單胞菌(Pseudomonas)等多種細菌感染時可通過鞭毛蛋白激活NLRC4炎癥小體組裝[12]。
2.3 菌體的內部物質(核酸、質粒)細菌內部物質中的核酸(dsDNA、RNA)、質粒可激活AIM2、NLRP3、NLRP1炎癥小體。AIM2蛋白可識別單核細胞增生李斯特菌(Listeria monocytogenes)、弗朗西斯氏菌(Francisella)、肺炎鏈球菌(Streptococcus pneu?moniae)和牛布魯氏菌(Brucella abortus)細菌胞質dsDNA激活AIM2炎癥小體組裝[13]。此外細菌RNA可激活NLRP3炎癥小體組裝,如B族鏈球菌提取的純化總RNA、大腸桿菌的mRNA和RNA-DNA雜交體、結核分枝桿菌中的dsRNA均可激活NLRP3炎癥小體[14-17]。進一步研究表明,細菌mRNA、tRNA和
rRNA也能激活人巨噬細胞的NLRP3炎癥小體[18]。除細菌核酸外,最新研究發現細菌質粒也可激活NLRP1炎癥小體,福氏志賀菌分泌的侵襲性質粒抗原H7.8(IpaH7.8)是一種可誘導NLRP1B降解和激活的蛋白酶,可激活NLRP1炎癥小體引發經典細胞焦亡途徑[19-20]。
近年來,黑社會性質組織向政治領域滲透的現象又出現了新的趨勢:一是通過暴力、威脅、賄賂等手段,操縱選舉,直接控制農村基層政權;二是通過收買有權勢的上級領導,控制下屬組織謀取暴利。[15]
2.4 細菌分泌系統效應蛋白細菌通用的Ⅰ~Ⅵ型細菌分泌系統中,可激活炎癥小體組裝的分泌系統包括Ⅲ型、Ⅵ型分泌系統(T3SS、T6SS)。研究發現,大腸桿菌、志賀氏菌(Shigella)、耶爾森菌(Yersinia)等多種細菌感染可通過T3SS桿狀蛋白和針狀蛋白激活NLRC4炎癥小體[12]。此外來自副溶血弧菌(Vibrio parahaemolyticus)的T3SS效應蛋白VopS及新洋蔥伯克霍爾德菌(Burkholderia cenocepacia)的T6SS效應蛋白TecA可激活Pyrin炎癥小體[21-22]。除通用的Ⅰ~Ⅵ型分泌系統,分枝桿菌屬中存在特有的Ⅶ型分泌系統(ESX-1型分泌系統),可激活NLRP3炎癥小體,如結核分枝桿菌(Mycobacterium tuberculosis)Ⅶ型分泌系統中早期分泌抗原靶蛋白ESAT-6(6 kD early secretory antigenic target protein)[11]。同時海洋分枝桿菌(Mycobacterium marinum)的ESX-1型分泌系統也可激活NLRP3炎癥小體,但并未找到具體誘導蛋白[23]。
2.5 細菌的分泌外毒素根據作用于靶細胞的方式不同,外毒素主要分為三類:作用于細胞膜毒的素、損傷細胞膜的毒素和細胞內作用毒素。這些細胞毒素中,可激活炎癥小體毒素主要包括損傷細胞膜的毒素和細胞內作用毒素。
2.5.1 損傷細胞膜毒素損傷細胞膜毒素在體外具有溶血素或溶細胞素活性,主要包括孔形成毒素和磷脂酶類毒素,兩者均可激活NLRP3炎癥小體。如孔形成毒素中金黃色葡萄球菌分泌的α-、γ-溶血素、殺白細胞素、殺白細胞素A/B[24];也包括弧菌屬中霍亂弧菌(Vibrio cholerae)和創傷弧菌(Vibrio vul?nificus)分泌的溶血素和MARTX毒素、副溶血性弧菌(Vibrio parahaemolyticus)分泌的耐熱直接溶血素、河流弧菌(Vibrio fluvialis)分泌的溶血素、蛋白水解弧菌(Vibrio proteolyticus)分泌的殺白細胞素[25-26]。磷脂酶類毒素包括金黃色葡萄球菌β溶血素等。
2.5.2 細胞內作用毒素細胞內作用毒素主要具有酶活性,通過酶活性影響細胞內生物機制或抑制蛋白合成,包括多種腺苷酸環化酶、ADP-核糖轉移酶、葡(萄)糖基轉移酶、神經毒素等毒素,主要激活NLRP3和Pyrin炎癥小體。如百日咳鮑特菌分泌的百日咳腺苷酸環化酶毒素(adenylate cyclase toxin,CyaA)可激活NLRP3炎癥小體,同時其分泌的百日咳毒素(pertussis toxin,PTX)依賴其ADP核糖基轉移酶可激活Pyrin炎癥小體誘導小鼠腹腔細胞IL-1β分泌[27-28]。同時化膿鏈球菌分泌的SpyA和銅綠假單胞菌分泌的PEA兩種ADP-核糖基轉移酶毒素(ADP-ribosyltransferase toxin)也可激活體內細胞NLRP3炎癥小體[29]。此外艱難梭菌(Clostridium difficile)分泌的艱難梭菌毒素A(Tcd A)和艱難梭菌毒素B(Tcd B)兩種葡糖基轉移酶毒素既可激活NLRP3炎癥小體也可激活Pyrin炎癥小體[30]。
綜上,細菌感染時可通過其自身不同組件激活體內多種炎癥小體介導炎癥反應。具體來說,細菌鞭毛和T3SS效應蛋白可激活NLRC4炎癥小體,細菌分泌的成孔毒素、酶類毒素、某些效應蛋白、RNA可激活NLRP3炎癥小體,細菌dsDNA可激活AIM2炎癥小體,細菌分泌的某些毒素可激活Pyrin炎癥小體,同時細菌的LPS可激活非經典炎癥小體途徑,引發細胞焦亡和細胞因子IL-1β、IL-18成熟與分泌。
3 細菌感染激活炎癥小體介導細胞焦亡的機制研究
細菌通過不同組件激活不同炎癥小體誘導細胞焦亡,各炎癥小體激活過程中涉及機制也不同,主要分為直接激活與間接激活。直接激活中,主要激活機制為不同蛋白結構的直接切割與結合。如細菌的效應物質可切割NLRP1蛋白激活炎癥小體組裝,細菌的鞭毛蛋白和III型分泌系統效應蛋白結合NAIP、dsDNA結合AIM2分別激活NLRC4、AIM2炎癥小體介導經典細胞焦亡途徑,LPS結合caspase-4/5/11直接介導非經典細胞焦亡途徑。間接激活中,細菌組件擾動細胞內信號進而激活Pyrin和NLRP3炎癥小體。以下將針對每一炎癥小體介紹其蛋白結構進而闡述其激活機制。
3.1 直接激活
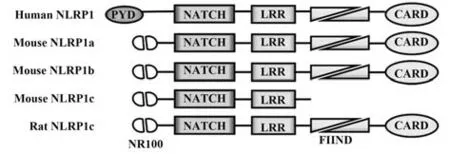
圖1 人、小鼠、大鼠NLRP1蛋白結構[31]Fig.1 NLRP1 protein structure of human,mouse and rat[31]
目前研究確定的可激活NLRP1炎癥小體的細菌效應物質包括炭疽桿菌分泌的炭疽致死毒素(lethal toxin,LT)的致死因子(lethal factor,LF)及福氏志賀菌分泌的IpaH7.8,其激活機制均在于可直接切割或酶解NLRP1蛋白的N末端,釋放其C端片段進而激活caspase-1[19-20]。綜上,NLRP1激活的先決條件為FIIND的自蛋白分解、對刺激的響應和N-末端蛋白分解,進而指出細菌感染主要通過其組件切割NLRP1蛋白激活NLRP1炎癥小體介導細胞焦亡。
3.1.2 細菌感染結合NAIP激活NLRC4炎癥小體引發細胞焦亡NLRC4炎癥小體是目前已知較為清楚的炎癥小體之一,其蛋白由3部分組成,N端CARD結構域、中間NACTH結構域和C端LRR結構域。NLRC4通過直接的NLRC4CARD-caspase-1CARD相互作用激活caspase-1。
多種細菌感染可通過鞭毛蛋白和T3SS效應蛋白激活NLRC4炎癥小體。分析其激活機制發現,與其他炎癥小體不同,NLRC4激活需要另一種蛋白,即神經元凋亡抑制蛋白(NAIPs),可在胞漿中識別細菌成分的存在。沒有這些細菌組分存在時,NAIP和NLRC4中的LRR結構域與其自身NBD相互作用維持自抑制狀態[34]。細菌配體存在的情況下,NAIP直接識別并結合特定配體,從而打破其自抑制構象,允許其NBD介導與NLRC4的共齊聚[35]。這種共齊聚反應誘導形成一個大的NAIP-NLRC4炎癥小體,激活caspase-1,最終導致IL-1β/IL-18釋放和細胞焦亡。NAIP蛋白是NLR家族成員之一,小鼠有4種NAIP同源基因:NAIP5/NAIP6識別鞭毛蛋白;NAIP1和NAIP2分別特異性識別T3SS針狀蛋白和桿狀蛋白。而人類僅編碼1個NAIP基因(hNAIP),T3SS桿狀/針狀蛋白和鞭毛蛋白均能被識別[12]。目前研究表明,NLRC4的Ser533磷酸化激活了NLRC4,用于通過NAIP感應配體的后續激活[36]。但也有與之相矛盾的報道表明,這種磷酸化也發生在NLRC4的非活性狀態。因此,需要進一步研究確定NLRC4-Ser533磷酸化的必要性。
3.1.3 細菌感染結合AIM2激活AIM2炎癥小體引發細胞焦亡相對于NLRC4炎癥小體激活時結合NAIP才可引發pro-caspase-1聚集,AIM2炎癥小體的激活機制更為簡單,可通過本身HIN200結構域直接結合細菌胞質dsDNA誘導炎癥小體組裝引發細胞焦亡。AIM2能與胞質細菌雙鏈DNA(dsDNA)相互作用使ASC和pro-caspase-1結合形成功能性炎癥小體。
靜息細胞中,AIM2中的PYD通過靜電吸引結合其HIN200結構域,使AIM2持續處于自抑制狀態。當細菌感染時,細菌的dsDNA對HIN200結構具有更高的親和力。因此,細胞內dsDNA出現后,PYD被dsDNA取代,從而使AIM2取消自抑制狀態[37]。進一步,PYD暴露于ASC,通過同型PYDPYD相互作用招募ASC。隨后通過ASC-CARD和procaspase-1-CARD結合組裝成一個功能性AIM2炎癥小體,引發炎癥反應和細胞焦亡。
dsDNA-AIM2炎癥小體途徑是宿主細胞抵御細菌和病毒病原體的重要途徑,涉及多種信號通路,包括AIM2直接識別細菌DNA,也包括胞漿雙鏈DNA激活IFN刺激DNA[interferon(IFN)-stimulatory DNA,ISD]途徑,即cGAS-STING-TBK1-IRF3途徑促進AIM2感知dsDNA。ISD途徑中,環GMP-AMP合酶(cGAS)獨立于其序列感知DNA,激活胞質DNA傳感適配器-IFN基因刺激因子(stimulator of interferon genes,STING)。STING隨后激活蛋白激酶IKK和TBK1,進而激活轉錄因子NF-κB和干擾素調節因子3(IRF3),誘導Ⅰ型IFN產生,Ⅰ型IFN進一步通過細胞膜IFNAR1受體進入細胞參與AIM2感知dsDNA。此外,不同細菌感染涉及機制略有差別,如肺炎鏈球菌與單核細胞增生李斯特菌感染時肺炎鏈球菌溶血素(pneumolysin,PLY)、李斯特菌溶素O(listeriolysinO,LLO)均可激活AIM2炎癥小體。弗朗西斯氏菌激活AIM2炎癥小體時涉及多種機制,包括細菌內化、溶酶體酸化、線粒體ROS產生、K+外流、鳥苷酸結合蛋白(GBPs)和IFN誘導蛋白IRGB10參與[13]。
3.2 間接激活
3.2.1 細菌感染失活Rho-GTPases激活Pyrin炎癥小體引發細胞焦亡與直接激活中蛋白間結構切割與結合不同,間接激活主要通過擾動細胞內信號進而激活炎癥小體引發經典細胞焦亡途徑,主要包括Pyrin、NLRP3兩種炎癥小體。
研究表明,在靜息細胞中,Pyrin作為不活躍的蛋白存在,其B-Box結構域與PYD、CC結構域自齊聚成三聚體,維持Pyrin自抑制狀態。細菌感染可通過毒素或效應蛋白翻譯后修飾Rho-GTPase的開關Ⅰ區使其失活,進而被Pyrin識別,觸發Pyrin炎癥小體激活[38]。如艱難梭菌分泌的細胞毒素TcdA和TcdB可引起Rho-GTPase家族成員中的RhoA糖基化[39];肉毒梭菌C3毒素可引起RhoA去酰胺化[38];副溶血性弧菌T3SS效應蛋白VopS、睡眠嗜組織菌IbpA蛋白均可引發RhoA FIC-結構域腺苷酸化[38];新洋蔥伯克霍爾菌T6SS效應器TecA引發RhoA去酰胺化[21]。此外,目前也發現百日咳鮑特菌分泌的PTX毒素依賴其ADP核糖基轉移酶活性誘導小鼠腹腔細胞IL-1β分泌,但未研究PTX與Rho-GTPase的關系[28]。這些翻譯后修飾均使Rho-GTPase失活。進一步分析翻譯后修飾的Rho-GTPase與Pyrin關系發現,Pyrin似乎未直接與修飾的Rho-GTPase結合,而是引起Pyrin去磷酸化和14-3-3蛋白解離。研究發現,靜息狀態下,Rho-GTPases激活了絲氨酸-蘇氨酸激酶PKN1和PKN2,在S208和S242結合并磷酸化Pyrin蛋白。磷酸化的Pyrin與14-3-3蛋白結合維持Pyrin炎癥小體非活性狀態。但當Rho-GTPase被細菌毒素或效應蛋白修飾時,Pyrin去磷酸化和14-3-3蛋白解離導致構象變化,從而使Pyrin炎癥小體活化[40-41]。但Pyrin如何去磷酸化以及14-3-3蛋白如何釋放尚未闡明。
3.2.2 細菌感染激活NLRP3炎癥小體引發細胞焦亡NLRP3與其他NLRP家族蛋白一致,其N端具有PYD。與其他NLRs相比,NLRP3在細胞靜息狀態下的表達太低不足以激活炎癥小體組裝。目前關于NLRP3炎癥小體激活的機制建立了雙信號模型。第一信號指可上調NLRP3和pro-IL-1β所需的啟動信號,TLR4、NOD2、TNFR和IL-1R等模式識別受體可激活啟動信號,這些信號可引起NF-κB激活,從而增加pro-IL-1β和NLRP3合成。第二信號被稱為感應信號,可由廣泛的結構不同的激動劑觸發,包括二氧化硅、石棉、鋁等環境晶體污染物、成孔毒素和核酸等病原體組件及血清淀粉樣蛋白A和ATP等內源性危險信號[42]。
多種細菌感染可激活人或小鼠細胞NLRP3炎癥小體,活化caspase-1,引發IL-1β和IL-18炎癥因子成熟與分泌或細胞焦亡。激活NLRP3炎癥小體的細菌組件包括成孔毒素或具有酶活性的毒素、某些效應蛋白和細菌RNA。如此眾多的激動劑如何激活細胞內NLRP3炎癥小體組裝,既往研究指出可能與鉀離子外流、線粒體DNA釋放、線粒體功能障礙、線粒體相關ROS產生、溶酶體相關組織蛋白酶B釋放、胞內鈣離子濃度改變、細胞膜孔道形成等相關,但尚未找出激活的統一通路。目前有幾項研究從細胞內的另外一些信號通路出發嘗試闡明其激活機制。一方面有研究指出NIMA相關激酶7(NIMA-related kinase 7,NEK7)可直接與巨噬細胞內NLRP3結合,是NLRP3炎癥小體組裝的核心驅動因子。NEK7的激酶結構域直接與NLRP3的LRR結構域結合形成復合物,與ASC和pro-caspase-1結合組裝后形成NLRP3炎癥小體復合物[43-45]。同時也有研究指出高爾基體反面網絡結構(trans-Golgi net?work,TGN)是一種新的共同細胞信號,研究表明多種差異極大的激動劑均能引起細胞內TGN解體為分散的特殊結構(dispersed TGN,dTGN)。dTGN形成后,其膜上富集的帶負電荷的磷脂酰肌醇-4-磷酸(phosphatidylinositol-4-phosphate,PtdIns4P)會招募原本處于細胞質基質內的NLRP3至dTGN,誘導ASC聚合,激活下游信號級聯[46-48]。但NLRP3炎癥小體的激活機制仍需進一步驗證。
綜上,細菌組件激活炎癥小體時既可通過不同蛋白結構間的切割或結合直接組裝炎癥小體,也可擾動細胞通用信號進一步活化炎癥小體組裝進一步促進下游炎癥反應發生。同時直接激活與間接激活間也存在聯系,如AIM2炎癥小體激活既可直接結合dsDNA結構直接激活也可擾動細胞信號通路間接激活。
4 總結
綜上,細菌感染宿主時,可通過其本身不同組件、不同機制激活宿主細胞體內多種炎癥小體,引發IL-1β、IL-18成熟與釋放及細胞焦亡。細胞分泌IL-1β、IL-18和誘導細胞焦亡發揮誘發、放大和持續炎癥反應的作用以及在多數情況下可幫助宿主抵御病原體感染。同時炎癥小體激活后也會對機體產生一些影響,如誘發凝血級聯反應及增強局部血管通透性[48-49]。
盡管目前對炎癥小體激活機制和效應反應有了大量研究進展,但對炎癥小體對抗微生物感染的認識還十分有限,仍存在諸多問題。如在炎癥小體激活機制中,翻譯后修飾起的具體作用是什么?炎癥小體激活與其他細胞死亡途徑(凋亡、壞死、自噬)是否存在其他聯系?同時在炎癥小體效應反應中,炎癥反應與細胞焦亡的作用是否可幫助清除體內感染的病原體?炎癥小體組裝與炎癥性脂質、抗菌遞質產生、細胞遷移和適應性免疫等其他細胞生理過程間的聯系為何?炎癥小體抑制劑是否可用于膿毒癥、自身免疫性疾病的臨床治療?對這些問題的理解將為宿主與病原菌的相互抗衡作用研究提供進一步見解,以便更好地理解細菌感染與宿主免疫防御間的博弈戰。
