多產芳烴并兼產小分子烷烴的正己烷轉化機理
2023-02-02 07:24:58吳冰峰于中偉王子健王麗新馬愛增代振宇
石油學報(石油加工) 2023年1期
吳冰峰, 于中偉, 王子健, 王麗新, 馬愛增, 代振宇
(中石化石油化工科學研究院有限公司,北京 100083)
石油化工過程中會副產大量輕烴資源,如裂解C5餾分、芳烴抽余油、重整拔頭油等。如何合理有效地利用這些資源,對于提高煉油企業的經濟效益至關重要。近年來,以低價值輕烴作為原料的輕烴芳構化技術受到了廣泛關注,其產品主要為混合芳烴和以低碳烷烴為主的液化氣組分。然而,受困于錯綜復雜的反應網絡,對于輕烴轉化機理的深入理解一直是一個挑戰[1]。
輕烴芳構化反應的核心是催化劑,一般以非貴金屬改性的HZSM-5分子篩為主。Caeiro等[2]認為,丙烷在Ga/ZSM-5催化劑上的芳構化過程是Br?nsted(B)酸和Lewis(L)酸協同作用的結果,其中脫氫步驟均在L酸上完成,而如烯烴的齊聚和裂化,二烯烴的環化步驟則是在B酸上完成。忻睦迪等[3]研究了制備方法對Zn/ZSM-5上Zn物種賦存狀態的影響,結果表明,原位水熱合成及液相離子交換法可使Zn2+遷移至分子篩的陽離子位,機械混合法及固相離子交換法制備的樣品中,Zn物種主要以ZnO形式賦存在分子篩的外表面,而等體積浸漬法制備得到的樣品中,上述2種Zn物種同時存在,而且得益于雙功能之間良好的協同作用,等體積浸漬法制備的催化劑在正庚烷催化裂解中具有更高的苯(B)、甲苯(T)和二甲苯(X)選擇性。
分子模擬技術作為實驗研究的重要補充工具,能模擬反應過程中反應分子的原子級行為,從而幫助科研人員更好地理解反應機理,因此分子模擬已被廣泛地應用于輕烴芳構化機理的研究中[4-5]。Pidko等[5]通過密度泛函理論(Density functional theory, DFT)研究對比了[Zn-O-Zn]2+和不同位置的Zn2+活化乙烷的能力,計算結果表明,雖然乙烷在[Zn-O-Zn]2+活性位上發生C—H鍵的異裂相對較為容易,但后續H2的重組脫附需要相對更高的能量。Liu等[6]借助分子模擬證實了C—H鍵在[Zn-O-Zn]2+活性位上異裂后,形成的Zn-OH活性位具有一定的B酸強度,在該活性位的協助下可以完成環化和齊聚等反應。然而,目前對輕烴轉化機理的研究尚不系統,而且未對輕烴芳構化的產物分布做出合理的解釋。
因此,筆者以正己烷作為模型化合物,通過實驗和分子模擬相結合的方法,對輕烴轉化反應機理進行了系統研究,為提高輕烴芳構化技術升級提供理論依據。
1 實驗部分
1.1 原料和試劑
硝酸和正己烷,分析純,天津市大茂化學試劑廠產品;六水合硝酸鋅(Zn(NO3)2·6H2O),分析純,國藥集團化學試劑有限公司產品;HZSM-5分子篩、Silicalite-1(S-1)分子篩、田菁粉,中國石化石油化工科學研究院提供;Sasol Boehmite(SB)粉,Sasol公司產品。
1.2 催化劑制備
將100 g納米HZSM-5分子篩(n(SiO2)/n(Al2O3)=50)與等質量的SB粉充分混合,加入90 mL的硝酸水溶液混捏成塊狀,使用雙螺桿擠出機將其擠條成型,25 ℃下風干后,轉入120 ℃烘箱中干燥4 h,隨后在馬弗爐中550 ℃下煅燒4 h,最后破碎、過篩保留20~40目的樣品,并記為HZ-5。另外,將SB粉與少量田菁粉混合,并通過與上述相同的過程將其制成20~40目的條狀γ-Al2O3樣品。另將S-1分子篩通過壓片法,制成同樣大小的塊狀S-1以備后續使用。
采用等體積浸漬法制備金屬改性的催化劑。將HZ-5、γ-Al2O3和S-1樣品分別倒入Zn(NO3)2水溶液中充分攪拌,Zn(NO3)2水溶液的濃度根據負載量而定,浸漬后的材料在25 ℃下靜置4 h,隨后轉入120 ℃烘箱中干燥2 h,最后在550 ℃的馬弗爐中煅燒4 h。根據ZnO所占的質量分數,分別將所得樣品記為wZnO/HZ-5,wZnO/γ-Al2O3和wZnO/S-1(wZnO/HZ-5催化劑中w分別為1%、3%和5%,wZnO/γ-Al2O3和wZnO/S-1中w均為3%)。
1.3 催化劑表征
采用美國Micromeritics公司生產的Auto Chem II-2920型化學吸附儀對催化劑進行程序升溫氨脫附(NH3-TPD)測試。約0.1 g樣品,在He氣氛中加熱至500 ℃,然后降溫至150 ℃進行NH3吸附,隨后用He吹掃除去物理吸附的NH3,最后以升溫速率10 ℃/min升溫至700 ℃,并利用熱導檢測器(TCD)檢測脫附下來的NH3。
采用美國Nicolet公司生產的is10型傅里葉變換紅外光譜儀對催化劑樣品進行羥基-IR(OH-IR)測定,儀器分辨率為4.0 cm-1,掃描范圍為4000~400 cm-1,將研磨好的樣品壓成10 mg左右的自支撐薄片,在紅外池中升溫至400 ℃,并抽真空脫附4 h,在降至25 ℃后測定紅外羥基譜圖。
采用美國Nicolet公司生產的is10型傅里葉變換紅外光譜儀對催化劑樣品進行吡啶-IR(Py-IR)分析,將樣品在400 ℃、高真空下凈化處理4 h后冷卻至25 ℃吸附吡啶分子,待吸附飽和后,再升溫至200 ℃,真空脫氣0.5 h,測定紅外譜圖,隨后在350 ℃下,真空脫氣0.5 h,測定紅外譜圖。
采用美國Thermo Fisher Scientific公司生產的Escalab 250Xi型X射線光電子能譜儀對催化劑樣品進行X射線光電子能譜(XPS)表征,采用污染碳C 1s結合能(284.6 eV)為基準進行校正。
1.4 芳構化反應實驗方法
采用小型固定床反應器進行芳構化反應實驗,其裝置示意圖如圖1所示。
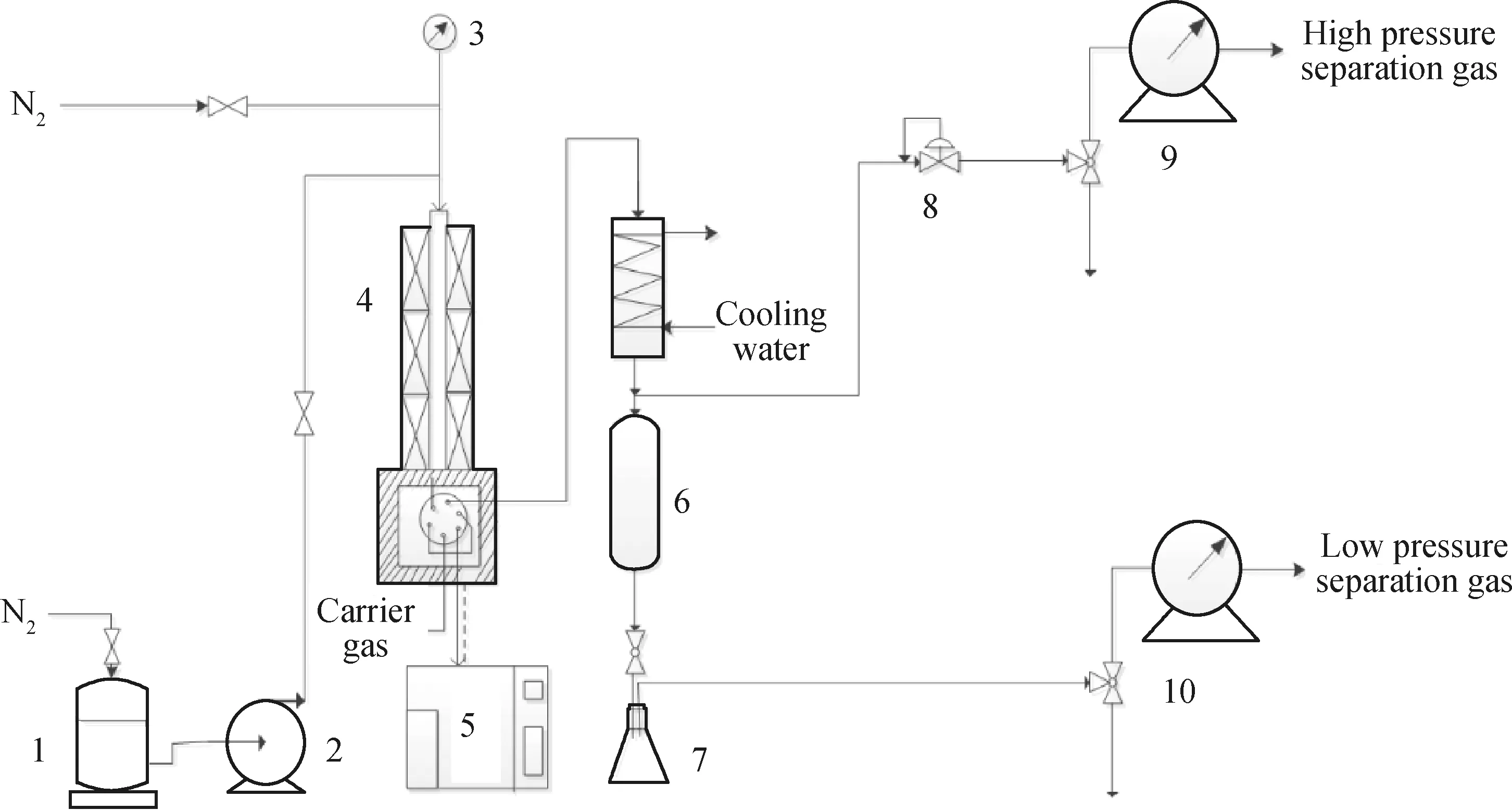
1—Storage tank; 2—Pump; 3—Pressure gage; 4—Reactor; 5—Gas chromatograph; 6—High pressure separation tank; 7—Liquid product tank; 8—Counterbalance valve; 9,10—Wet test meter圖1 芳構化反應實驗裝置示意圖Fig.1 Schematic diagram of experimental device for aromatization reaction
原料油通過壓罐法送入加熱的不銹鋼管式等溫反應器進行反應,其流量由流量計控制,總進料量用電子秤利用減重法計量,催化劑置于反應器恒溫段中部,兩端用石英棉與石英砂隔開。通過調整新鮮催化劑的裝量和反應物的進料速率,對反應物在催化劑上的停留時間進行控制,并用催化劑質量/反應物單位小時的摩爾進料量(mcat/F)來表示,從而考察不同停留時間下的反應情況。反應前,催化劑在N2流中升溫至500 ℃活化1 h,然后切斷N2,開啟流量計到目標流量開始進油。待反應平穩后開始計時,反應時間約10 h,每1.5 h進行在線取樣,采用美國Agilent公司生產的7890 B型氣相色譜儀進行在線分析,進樣口溫度為250 ℃,柱溫初始溫度為30 ℃,升溫速率為5 ℃/min,終點溫度為200 ℃,載氣為高純N2。待反應結束后,停止進油并關閉溫控系統,切換N2進行吹掃降溫,待降至25 ℃后,重新裝填新鮮催化劑考察下一停留時間的反應情況。
正己烷轉化率(x)、產物選擇性(s)和氫轉移指數(HTC)的計算式如式(1)~式(4)所示。
(1)
(2)
(3)
(4)
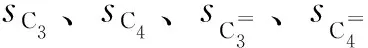
2 模型和計算方法
所有計算都基于BIOVIA公司Material Studio 8.0軟件的DMol3模塊完成。計算中所用泛函為GGA-PBE,基組為DNP。使用LST/QST方法對每個基元反應的過渡態進行搜索,并保證過渡態具有唯一的對應反應方向的虛頻。計算精度方面,能量、梯度和位移的收斂閾值分別為1.0×10-5Ha、2.0×10-4Ha/nm和0.005 nm。模擬部分所用基礎模型截取自ZSM-5分子篩直筒孔道和正弦孔道交匯處(如圖2所示),其中T8和T12位置的Si原子由Al原子取代。整個模型最外側Si原子形成的懸鍵由H原子來飽和,Si—H鍵的方向與原Si—O鍵的方向保持一致,鍵長為0.146 nm,且在整個計算過程中,終端H原子保持固定,其他原子均保持馳豫。ZnO改性模型中2個Zn2+原子分別與T8和T12處的Al原子周圍的O原子成鍵,并通過O橋相連,構成[Zn-O-Zn]2+。由于模型中包含過渡金屬Zn,因此在計算過程中選用DFT Semi-core Pseudopots對內層電子進行處理。用于對比的酸性模型,則在Al原子周圍的O原子上連接H原子平衡電荷,以模擬B酸。另外,由于T12位置是最容易接觸反應物的位置,因此無論是酸性模型還是金屬改性模型,模擬主要在T12位置附近的活性位上完成[7]。
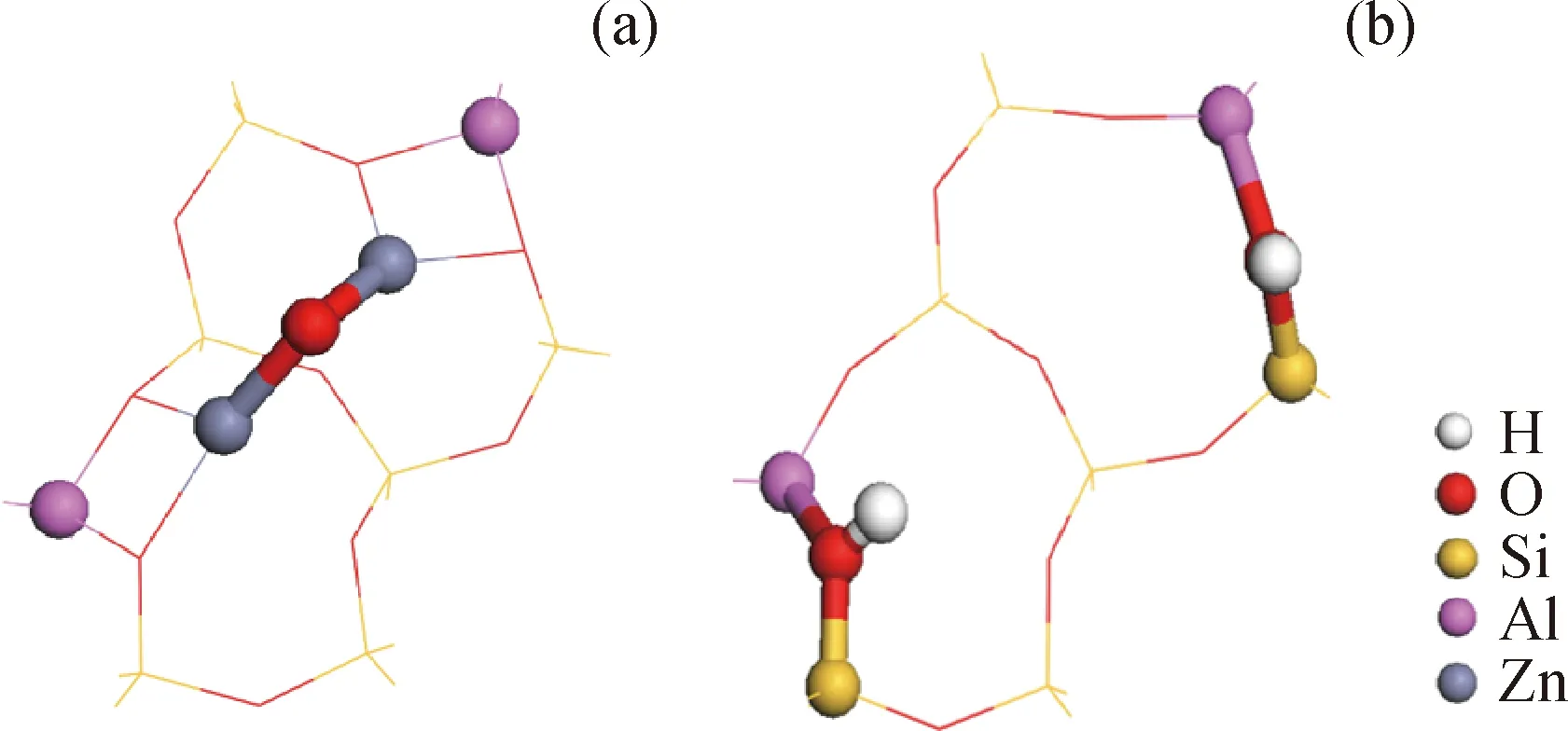
圖2 優化后的ZnO改性ZSM-5模型和HZSM-5模型Fig.2 Optimized ZnO modified ZSM-5 model and HZSM-5 model(a) ZnO modified ZSM-5 model; (b) HZSM-5 model
3 結果與討論
3.1 催化劑表征結果
3.1.1 OH-IR分析
為了解不同催化劑上ZnO物種的落位情況,首先對HZ-5、γ-Al2O3及ZnO/HZ-5系列催化劑進行了OH-IR表征,結果如圖3所示。圖3中出現在3770、3730、3726、3676和3603 cm-1處的紅外吸收峰分別對應Ⅰ6型鋁羥基、Ⅱ66型鋁羥基、缺陷處的硅羥基、Ⅲ型鋁羥基和硅鋁橋羥基[8-10]。對比圖3中各催化劑的譜圖可以發現,1%ZnO/HZ-5和3%ZnO/HZ-5催化劑的硅/鋁橋羥基的吸收峰強度相比于HZ-5催化劑出現了不同程度的減弱,3%ZnO/HZ-5的減弱程度相對較大,而其他羥基吸收峰強度減弱程度基本相同,這表明當ZnO負載量(質量分數)小于3%時,大部分ZnO物種被錨定在硅/鋁橋羥基上,少量分散于外表面缺陷處和Al2O3上。然而,當ZnO負載量提高到5%后,不僅硅/鋁橋羥基的吸收峰強度進一步減弱,而且表面缺陷處的硅羥基以及Ⅱ66型鋁羥基的吸收峰強度也出現了明顯的減弱,這表明此時ZnO物種除與硅/鋁橋羥基結合外,還有部分ZnO物種貯存于外表面的缺陷處和Al2O3表面上。
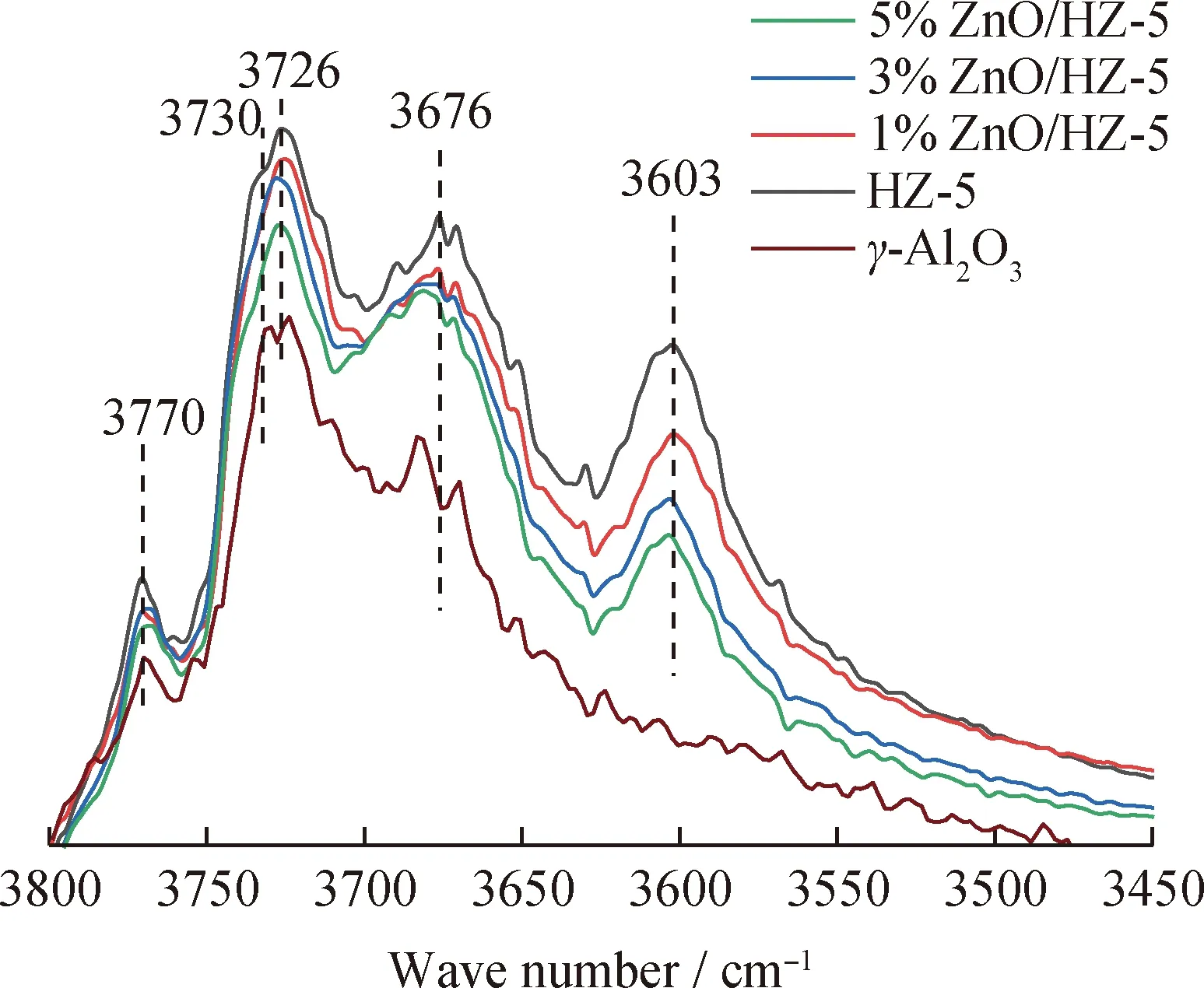
圖3 HZ-5、γ-Al2O3、1%ZnO/HZ-5、3%ZnO/HZ-5和5%ZnO/HZ-5催化劑的OH-IR譜圖Fig.3 OH-IR spectra of HZ-5, γ-Al2O3, 1%ZnO/HZ-5,3%ZnO/HZ-5 and 5%ZnO/HZ-5 catalysts
3.1.2 NH3-TPD和Py-IR分析
為了解ZnO含量對催化劑酸性和酸量的影響,對不同催化劑進行了NH3-TPD和Py-IR表征。圖4(a)為HZ-5、1%ZnO/HZ-5、3%ZnO/HZ-5和5%ZnO/HZ-5催化劑的NH3-TPD表征結果。從圖4(a)可以看出,隨著ZnO含量的增加,350~500 ℃范圍內的NH3脫附峰強度逐漸減小,而200~350 ℃范圍內的NH3脫附峰強度變化較小,這表明隨著ZnO負載量的增加,各催化劑上的強酸酸量持續減少,而弱酸酸量變化相對較小。圖4(b)為HZ-5、1%ZnO/HZ-5、3%ZnO/HZ-5和5%ZnO/HZ-5催化劑在200 ℃脫附后測定的吡啶吸附紅外光譜圖,B酸和L酸的定量數據列于表1。圖4(b)中1450 cm-1和1540 cm-1處的紅外吸收峰分別對應L酸和B酸,而1490 cm-1處的紅外吸收峰是B酸和L酸共同作用的結果。對比圖4(b)中紅外吸收峰的強度變化能夠發現,隨著ZnO含量的增加,B酸酸量逐漸減小,而L酸酸量逐漸增加。進一步結合表1中的數據可知,增加的L酸顯示出較強的酸性,這表明ZnO物種被錨定在強B酸上后,會使強B酸轉化為具有中等強度的L酸[6, 11]。值得注意的是,在ZnO/HZ-5系列催化劑的譜圖中,1613 cm-1處還出現了一肩峰,其強度隨ZnO含量的增加而增強,并且該峰并未在HZ-5的譜圖中觀察到,顯然該峰與ZnO物種有關。尹雙鳳等[12-13]在實驗中也觀察到了該峰的存在,并通過對比指出該峰是由吡啶在Zn-L酸上的吸附引起的。另外,圖4(b)還給出了羥基部分的譜圖,該范圍內僅在3720 cm-1和3680 cm-1處出現了對應硅羥基和Ⅲ型鋁羥基的紅外吸收峰,這主要是因為硅羥基和Ⅲ型鋁羥基的酸性相對較弱,吡啶分子與這2種羥基之間的相互作用較弱,因此脫附溫度較低,而吸附在硅/鋁橋羥基上的吡啶分子需400 ℃以上的脫附溫度[14]。此外,對比這兩處的紅外吸收峰強度變化能夠發現,3720 cm-1處的紅外吸收峰強度隨ZnO含量的增加而逐漸減弱,而3680 cm-1處的紅外吸收峰強度變化較小,這表明與Ⅲ型鋁羥基相比,ZnO物種更容易錨定在分子篩表面缺陷處的硅羥基上。
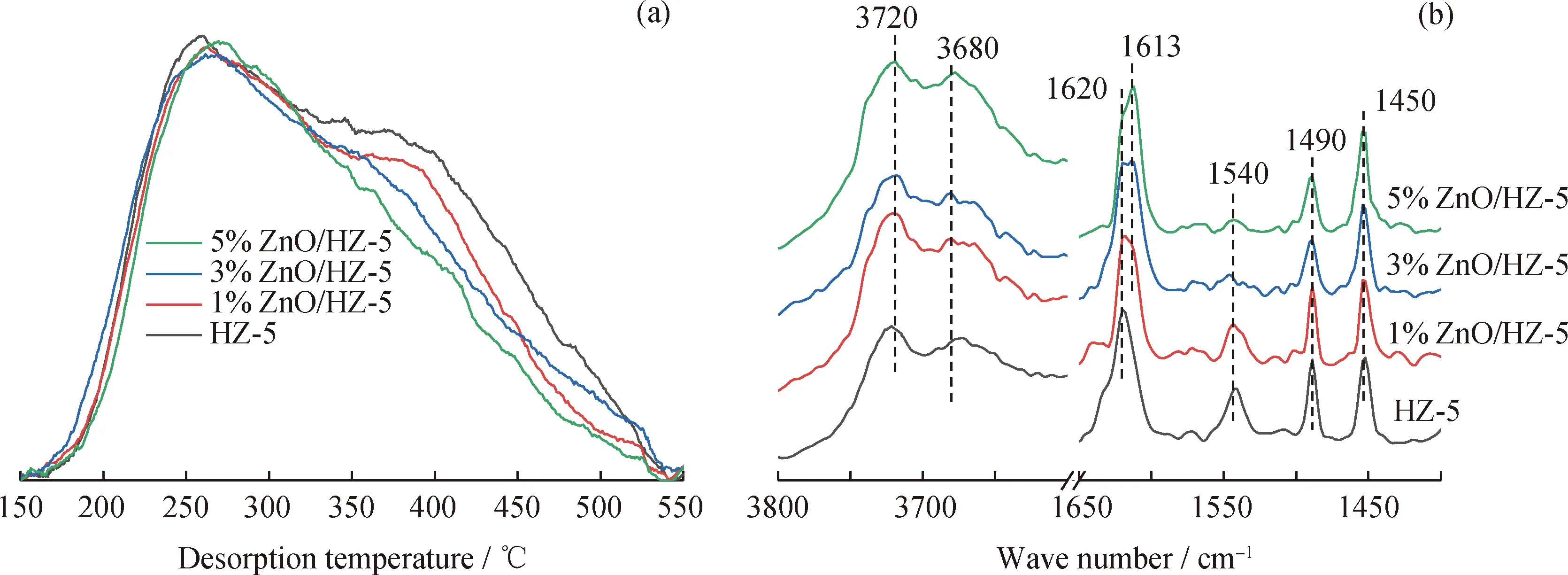
圖4 HZ-5、1%ZnO/Z-5、3%ZnO/Z-5和5%ZnO/Z-5催化劑樣品的NH3-TPD譜圖和Py-IR譜圖Fig.4 NH3-TPD and Py-IR spectra of HZ-5, 1%ZnO/Z-5, 3%ZnO/Z-5 and 5%ZnO/Z-5 catalyst samples(a) NH3-TPD spectra; (b) Py-IR spectra (Desorption at 200 ℃)
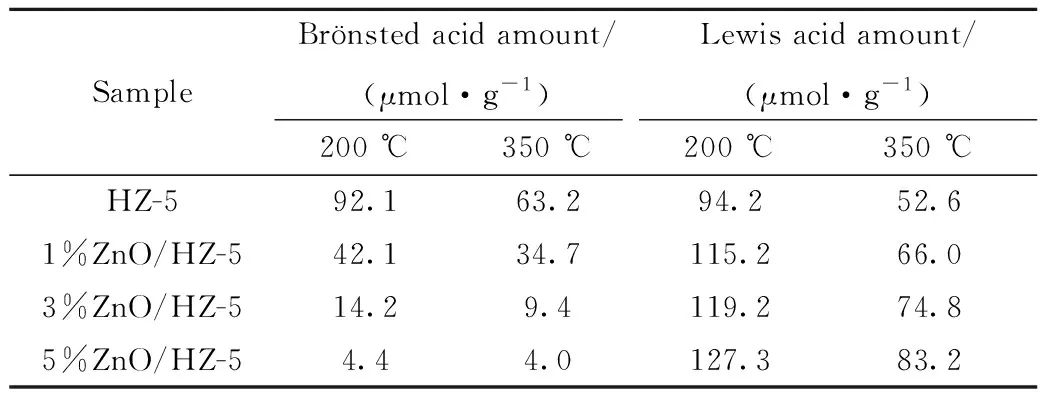
表1 HZ-5、1%ZnO/HZ-5、3%ZnO/HZ-5和5%ZnO/HZ-5催化劑樣品的Br?nsted酸量和Lewis酸量Table 1 Br?nsted and Lewis acid amounts of HZ-5,1%ZnO/HZ-5, 3%ZnO/HZ-5 and 5%ZnO/HZ-5 catalyst samples
3.1.3 XPS分析
XPS表征是分析催化劑表面金屬分散性與狀態的有效方法。圖5為ZnO/HZ-5催化劑上Zn(2p3/2)的XPS譜圖。經過解疊處理后得到處于1023 eV附近的譜峰α和1022 eV附近的譜峰β,其分別歸屬于[Zn-O-Zn]2+團簇和ZnO顆粒[15-16]。另外,圖5中還標出了兩譜峰的積分面積比(A(α)/A(β))。從圖5中數據可知,隨著ZnO負載量的提高,A(α)/A(β)持續下降,這表明隨著ZnO負載量的增加,ZnO物種的分散性逐漸變差,1%ZnO/HZ-5表面ZnO物種主要以[Zn-O-Zn]2+團簇形式存在,而3%ZnO/HZ-5和5%ZnO/HZ-5表面[Zn-O-Zn]2+和ZnO顆粒共存,且ZnO顆粒所占的比例逐步提高。另外,結合ZnO物種分散性的變化以及Py-IR表征結果,1613 cm-1處的紅外吸收峰可能是由吡啶分子在ZnO顆粒上的吸附所引起。
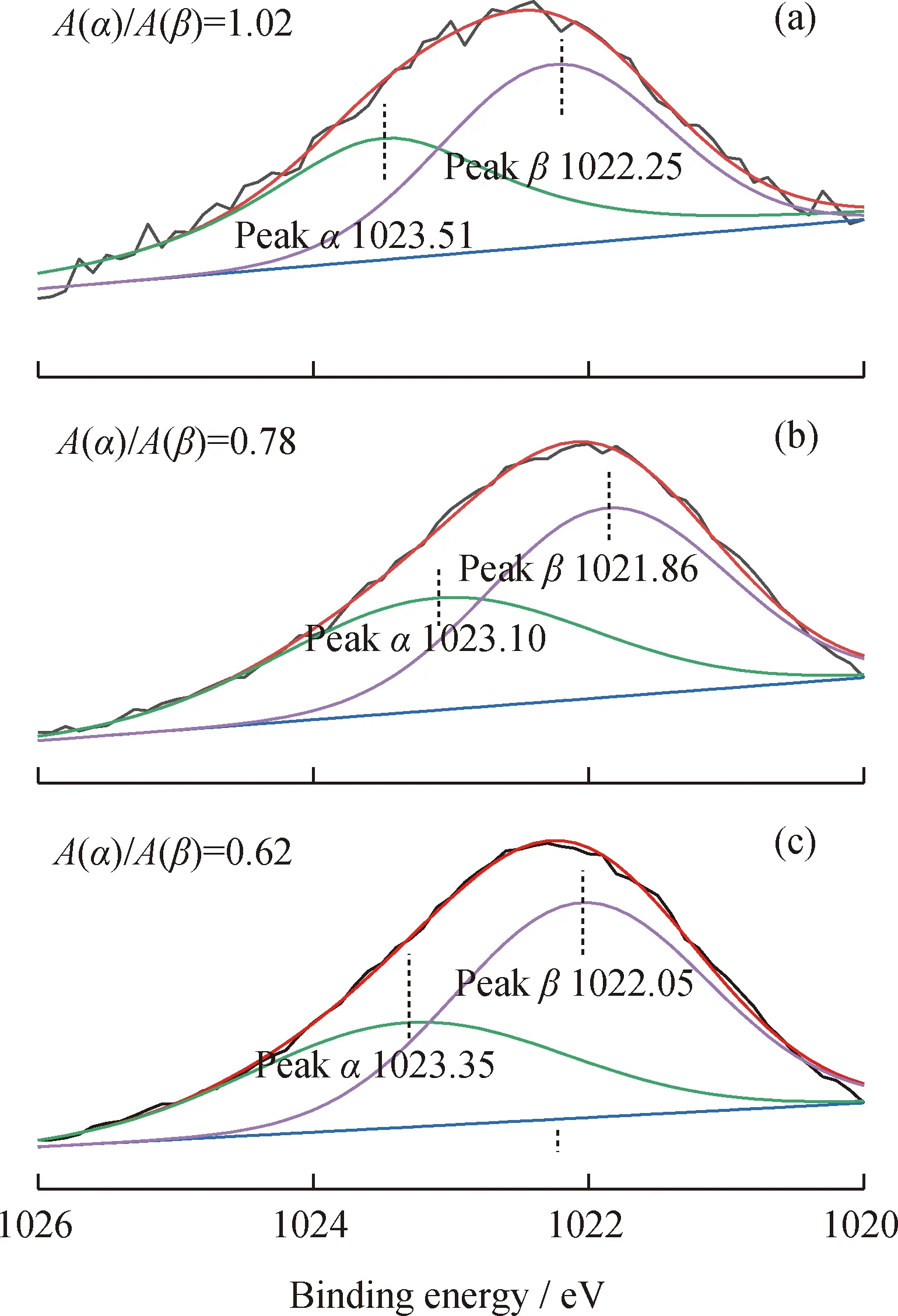
Raw data; Baseline; Zn2+ in the [Zn-O-Zn]2+ cluster; Zn2+ in the ZnO particle; Fitted curve圖5 1%Zn/HZ-5、3%ZnO/HZ-5和5%ZnO/HZ-5催化劑上Zn(2p3/2)的XPS譜圖Fig.5 XPS spectra of Zn(2p3/2) on 1%Zn/HZ-5,3%ZnO/HZ-5 and 5%ZnO/HZ-5 catalysts(a) 1%Zn/HZ-5; (b) 3%ZnO/HZ-5; (c) 5%ZnO/HZ-5
3.2 正己烷芳構化反應機理研究
3.2.1 正己烷芳構化實驗
表2為不同催化劑對正己烷芳構化反應的催化性能。從表2可以看出,分別以S-1和γ-Al2O3作為載體制備的3%ZnO/S-1和3%ZnO/γ-Al2O3的催化性能較差,而ZnO/HZ-5系列催化劑的催化性能明顯要更優,這表明主要的活性位來自于ZnO/HZ-5上的B酸和分子篩上的ZnO物種。對比發現,引入ZnO物種后,雖然烯烴選擇性有所下降,但H2以及芳烴的選擇性得到了明顯提高,丙烷選擇性大幅降低,這表明引入ZnO物種后,氫轉移反應得到抑制,催化劑的脫氫能力得到大幅增強。盡管如此,進一步對比1%ZnO/HZ-5,3%ZnO/HZ-5及5%ZnO/HZ-5之間的催化性能可以發現,ZnO負載質量分數超過3%后,H2和芳烴選擇性略有下降,但乙烷的選擇性有所增加。結合Py-IR和XPS的表征結果,這種實驗現象可能歸因于酸性和ZnO物種分散性的差異。對于1%ZnO/HZ-5,其上仍保留有較多的B酸,且氫轉移程度相對較強(HTC=27.56),丙烷選擇性相對較高;而5%ZnO/HZ-5上僅存少量的B酸,絕大部分脫氫反應在金屬位上完成,氫轉移程度較弱(HTC=17.00),并且由于ZnO含量較多,可能導致氫解反應,所以此時乙烷選擇性較高,丙烷和H2的選擇性較低[9,17]。顯然,為實現多產芳烴并兼產小分子烷烴的目的需要B酸和L酸之間良好的協同作用,因此ZnO負載量以3%為宜。
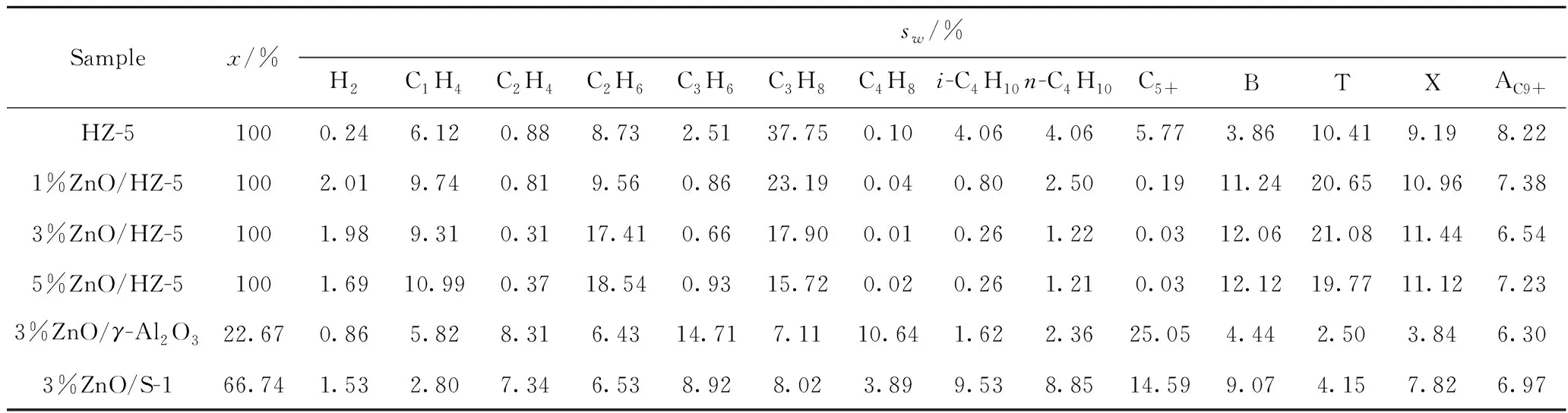
表2 不同催化劑對正己烷芳構化反應的催化性能對比Table 2 Comparison of catalytic performance of different catalysts for n-hexane aromatization
為探究正己烷在3%ZnO/HZ-5催化劑上的芳構化反應途徑,考察了不同停留時間下的反應情況(如圖6所示,細節數據列于表3)。由圖6可知,在轉化率小于58.02%時,H2的摩爾選擇性隨轉化率的升高而增大。結合表3數據可知,在轉化率小于58.02%時,H2與芳烴的摩爾選擇性之比約為3,并且該階段的HTC均小于3,顯然當停留時間mcat/F>8.60 (g·h)/mol時,芳構化途徑以脫氫反應為主。然而,當轉化率超過58.02%后,H2與芳烴的摩爾選擇性之比逐漸降低,而HTC大幅度提高,這表明當停留時間mcat/F>8.60 (g·h)/mol時,芳構化途徑以氫轉移為主。另外還發現,隨著停留時間的延長,丙烷選擇性持續降低,而甲烷和乙烷的選擇性卻快速提高,且二者幾乎相同,這說明當停留時間mcat/F>8.60 (g·h)/mol時,部分丙烷可能發生氫解反應生成甲烷和乙烷。
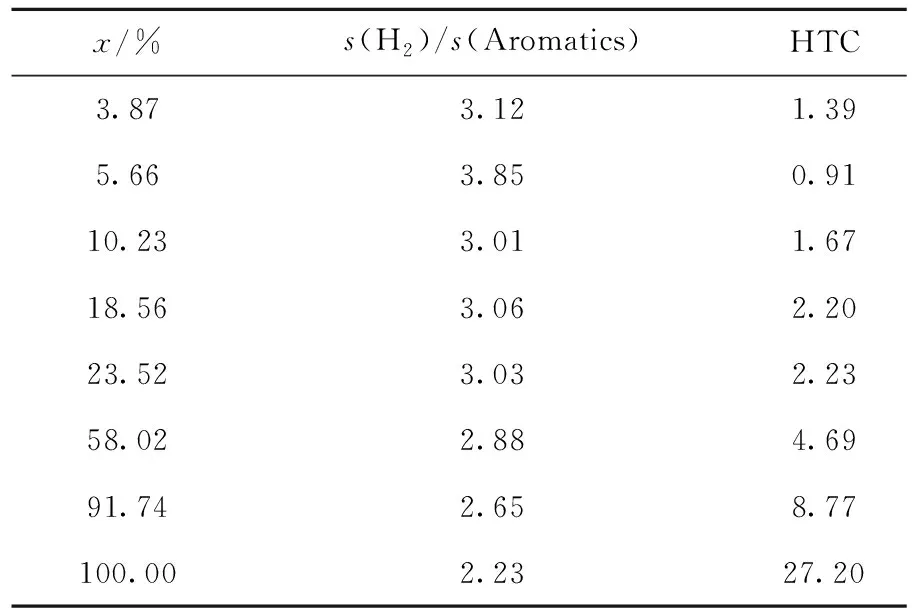
表3 不同正己烷轉化率(x)下H2與芳烴的摩爾選擇性(s)之比及HTCTable 3 Molar selectivity (s) ratio of H2 to aromatics and HTC at different conversion rates of n-hexane (x)
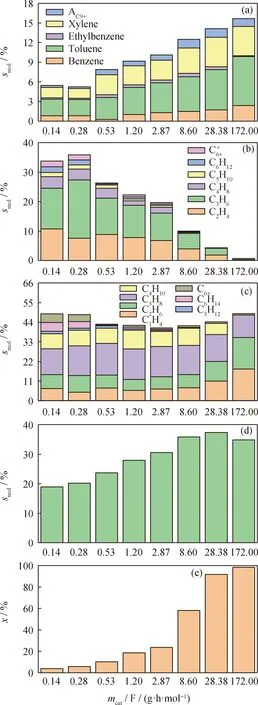
C6+—Alkanes with more than 6 carbon atoms; with more than 6 carbon atoms;AC9+—Aromatics with more than 9 carbon atoms圖6 停留時間(mcat/F)對正己烷在3%ZnO/HZ-5催化劑上芳構化反應的轉化率(x)和產物選擇性(smol)的影響Fig.6 Effect of residence time (mcat/F) on conversion rate (x) and product selectivity (smol) of the aromatization reaction of n-hexane on 3%ZnO/HZ-5 catalyst(a) Aromatic selectivity; (b) Alkene selectivity;(c) Alkane selectivity; (d) H2 selectivity; (e) Conversion of n-hexane
3.2.2 脫氫、氫轉移和氫解反應模擬研究
為探究實驗過程中脫氫途徑的變化,以正己烷的脫氫反應為例,通過分子模擬考察了正己烷在[Zn-O-Zn]2+活性位上的脫氫反應和在B酸性位上的氫轉移反應,結果如圖7所示。由圖7(a)可見,正己烷在[Zn-O-Zn]2+活性位上的脫氫反應可分為3步:(1)C—H鍵的異裂;(2)己烯的解離脫附;(3)H2的重組脫附。經計算,第二步己烯的解離脫附能壘最高,約為153.91 kJ/mol,因此該反應步驟為速控步驟,這一結論與文獻[18-19]的結果相一致。由圖7(c)可見,正己烷與丙烯之間的氫轉移反應過程可分為2步:(1)兩者形成具有H橋結構的中間體;(2)該中間體的分解。經過計算,該反應過程中具有H橋結構中間體的形成是速控步驟,其能壘約157.76 kJ/mol。很明顯,同種反應物在[Zn-O-Zn]2+活性位上的脫氫反應在動力學上更具優勢,因此當停留時間mcat/F<8.60 (g·h)/mol時,在金屬活性位上的脫氫反應較明顯,而mcat/F>8.60 (g·h)/mol時,氫轉移反應增多。
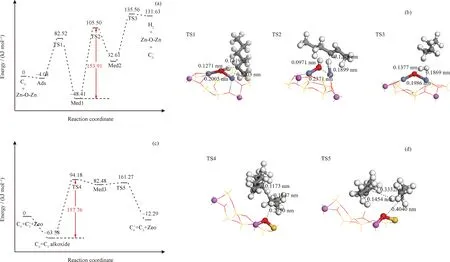
Ads—Adsorbed state; TS1, TS2, TS-3—Transition state of dehydrogenation reaction of n-hexane; Med1, Med2—Medium of dehydrogenation reaction of n-hexane; TS4, TS5—Transition state of hydride transfer reaction of n-hexane and propane; Med3—Medium of hydride transfer reaction n-hexane and propane; Zeo—Zeolite; Red ball—Oxygen; Yellow ball—Silicon; Pink ball—Aluminum; Grey ball—Carbon; White ball—Hydrogen圖7 正己烷在[Zn-O-Zn]2+活性位上的脫氫反應的勢能面及其過渡態結構和正己烷在Br?nsted酸位上的氫轉移反應的勢能面及其過渡態結構Fig.7 Potential energy surface of the dehydrogenation reaction of n-hexane at [Zn-O-Zn]2+ active site and its transition state structure and potential energy surface of the hydrogen transfer reaction of n-hexane at Br?nsted acid site and its transition state structure(a) Potential energy surface of dehydrogenation reaction of n-hexane; (b) Transition states structure of dehydrogenation reaction of n-hexane; (c) Potential energy surface of hydride transfer reaction of n-hexane and propane; (d) Transition states structure of hydride transfer reaction of n-hexane and propane
另外,對于丙烷的氫解反應進行了模擬計算,結果如圖8所示。丙烷的氫解反應過程分為2步:(1)[Zn-H]+上的H-離子進攻C—C鍵形成甲烷和Zn-C2H5;(2)[Zn-O-Zn]2+活性位的恢復并形成乙烷。其中,第一步為該氫解反應的速控步驟,能壘約為289.62 kJ/mol。由此可見:丙烷的氫解反應在動力學上是一個較慢的反應,需在足夠長的停留時間內才可能發生,這與實驗中的觀察相一致;并且由于氫解反應發生在金屬活性位上,將與脫氫反應產生競爭,不利于脫氫反應的進行,因此這將導致芳構化過程中脫氫途徑的變化。
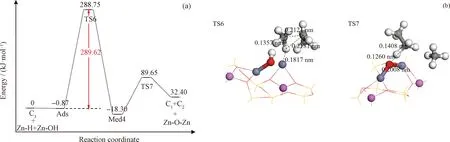
Ads—Adsorbed state; TS6, TS-7—Transition state of hydrogenolysis of propane; Med4—Medium of hydrogenolysis of propane; Red ball—Oxygen; Yellow ball—Silicon; Pink ball—Aluminum; Grey ball—Carbon; White ball—Hydrogen圖8 丙烷氫解反應的勢能面及其過渡態結構Fig.8 Potential energy surface of propane hydrogenolysis reaction and its transition state structure(a) Potential energy surface of propane hydrogenolysis reaction; (b) transition state structure of propane hydrogenolysis reaction
4 結 論
對多產芳烴兼產小分子烷烴的正己烷轉化機理進行了研究,主要得到以下結論:
(1)向HZ-5中引入適量的ZnO能夠削減B酸數量,抑制氫轉移反應,強化脫氫反應,提高芳烴選擇性,但過高的L酸酸量將加劇氫解反應,影響H2和芳烴選擇性,ZnO的負載質量分數以3%為宜。
(2)當正己烷在3%ZnO/HZ-5催化劑上的轉化率小于58.02%時,芳構化途徑以脫氫反應為主,H2和芳烴的摩爾選擇性有很好的對應關系;但在轉化率大于58.02%時,HTC快速增加,芳構化途徑以氫轉移反應為主。
(3)當停留時間mcat/F>8.60 (g·h)/mol時,甲烷和乙烷的選擇性快速上升,且二者的摩爾選擇性幾乎相同,甲烷和乙烷可能是丙烷氫解反應的產物。
(4)在[Zn-O-Zn]2+活性位上的脫氫反應能壘小于氫轉移反應能壘,并且丙烷的氫解反應能壘更高,因此氫解反應在停留時間mcat/F>8.60 (g·h)/mol時才能發生;而氫解反應的發生不利于脫氫反應的進行,因此導致脫氫途徑的變換。
猜你喜歡
課堂內外·初中版(科學少年)(2025年1期)2025-02-28 00:00:00
課堂內外·初中版(科學少年)(2025年2期)2025-02-28 00:00:00
英語世界(2023年10期)2023-11-17 09:18:18
科學大眾(中學)(2019年3期)2019-05-17 10:04:30
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
汽車觀察(2018年10期)2018-11-06 07:05:26
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
