Ni-Ce/N-rGO催化劑的表征及其催化苯酚加氫制備環己酮的性能
2023-02-02 07:45:08徐海升薛媚月黃國強郜鵬程
石油學報(石油加工) 2023年1期
關鍵詞:催化劑
徐海升, 薛媚月, 黃國強, 郜鵬程, 范 崢
(1.西安石油大學 化學化工學院,陜西 西安 710065;2.陜西省綠色低碳能源材料與過程工程技術研究中心,陜西 西安 710065; 3.西安市高碳資源低碳化利用重點實驗室,陜西 西安 710065)
隨著工業技術的快速發展,環境污染和能源短缺問題愈發嚴重[1-3]。生物質能源因具有清潔、可再生、來源廣等特點,受到越來越多的關注[4]。生物質中酚類化合物的含量相當豐富,尤其是苯酚,可選擇性加氫制備環己酮,且反應條件溫和、操作簡單、副反應少,應用前景廣闊[5]。苯酚加氫反應路徑為苯環先部分加氫生成環己烯醇中間體,環己烯醇極不穩定會快速異構化生成環己酮,環己酮繼續加氫則會生成環己醇[6]。在酸性催化劑上,苯酚以共平面方式與載體吸附,作用較強容易過度加氫生成環己醇;在堿性催化劑上,苯酚以非平面方式與載體吸附,產生的相互作用較小,有利于環己酮脫附進而避免了進一步加氫。
環己酮可用于生產己二酸和己內酰胺,還可在油漆、印刷等領域用作溶劑[7-8]。目前工業上常用環己烷氧化法和苯酚加氫法制備環己酮,前者反應需要在高溫高壓下進行,條件苛刻且副產物較多;后者反應條件相對溫和,但是催化性能較好的催化劑多為Pt、Pd、Ru等貴金屬,因此研究可以提高苯酚轉化率、環己酮選擇性且成本低廉的非貴金屬或雙金屬催化劑成為亟需解決的問題[9-12]。筆者所在課題組[13-15]先后進行了不同催化劑的探索性研究,發現Ni/N-rGO催化劑具有較好催化加氫性能且成本低,但Ni在高溫下容易團聚影響催化劑的活性。通過加入不同堿性組分Ce、Cu、Fe和Na對Ni/N-rGO催化劑進行改性,然后對催化劑表征,并考察其對苯酚選擇性加氫制備環己酮性能的影響。
1 實驗部分
1.1 原料和試劑
硝酸鈉、苯酚、環己酮,均為分析純,天津市科密歐化學試劑有限公司產品;鱗片石墨,分析純,安徽安特生物化學有限公司產品;尿素,分析純,天津永晟精細化工有限公司產品;硝酸鎳,分析純,天津市華東試劑廠產品;硝酸鈰,分析純,天津市福晨化學試劑廠產品;硝酸銅,分析純,國藥集團化學試劑有限公司產品;無水乙醇,分析純,天津市大茂化學試劑廠產品。
1.2 催化劑制備
采用水熱法制備N-rGO載體。稱取100 mg由改進Hummers法制備的氧化石墨(GO)溶于去離子水中,超聲處理2 h,得到1 mg/mL的氧化石墨烯分散液[16-17];然后緩慢把尿素加入GO分散液中(GO/尿素質量比為1/30),室溫下繼續超聲攪拌后將混合液轉移到水熱反應釜中,在180 ℃條件下水熱反應12 h,將反應釜中懸浮液離心,取下層固體用去離子水和無水乙醇反復抽濾洗滌至中性,然后于真空干燥箱充分干燥得到N-rGO載體。
采用浸漬法制備Ni-Cen/N-rGO催化劑(n表示Ce/Ni摩爾比n(Ce)/n(Ni),分別為0.1、0.2、0.3)。稱取一定量的N-rGO載體于燒杯中,按照不同n(Ce)/n(Ni)的要求加入0.1 mol/L的Ni(NO3)2·6H2O和Ce(NO3)3·6H2O水溶液,常溫下磁力攪拌2 h,浸漬24 h后在70 ℃水浴中恒溫攪拌蒸干水分,再轉移至80 ℃烘箱中充分干燥;然后在N2保護下于管式爐中400 ℃焙燒2 h,最后在H2氣氛下以升溫速率5 ℃/min升溫至450 ℃,停留還原2 h得到Ni-Cen/N-rGO催化劑。
1.3 催化劑表征
催化劑的比表面積(BET法)、平均孔徑和孔體積(BJH法)采用美國麥克公司生產的ASAP2020型物理吸附儀測定。催化劑的晶體結構采用德國Bruker公司生產的D8 ADVANCE型X射線衍射儀測定。催化劑的官能團結構采用美國熱電尼高力儀器公司生產的Nicolet Nuxes 670型紅外光譜儀測定。催化劑的形貌和元素分析采用美國FEI公司生產的Quanta 600 FEG型掃描電鏡及EDS能譜儀測定。
1.4 催化劑反應性能評價
取0.1 g催化劑和0.1 mol/L的苯酚水溶液30 mL填裝到100 mL間歇式高溫高壓反應釜中,通入H2,在反應溫度150 ℃、壓力0.4 MPa的條件下反應2 h,收集反應液并準確稱量質量和體積,利用UV-2600型紫外-可見光光度計測定苯酚和環己酮的吸光度,然后根據苯酚和環己酮的標準曲線計算收集液中苯酚和環己酮的物質的量,由式(1)和式(2)計算出苯酚轉化率(x,%)和環己酮選擇性(s,%)。
(1)
(2)
式(1)和式(2)中:m0為苯酚的初始質量,g;m1為苯酚的剩余質量,g;m2為反應后生成的環己酮質量,g。
2 結果與討論
2.1 催化劑表征結果
2.1.1 N2吸附-脫附表征
N-rGO載體及Ni-Cen/N-rGO催化劑樣品的比表面積和孔結構性質如表1所示。
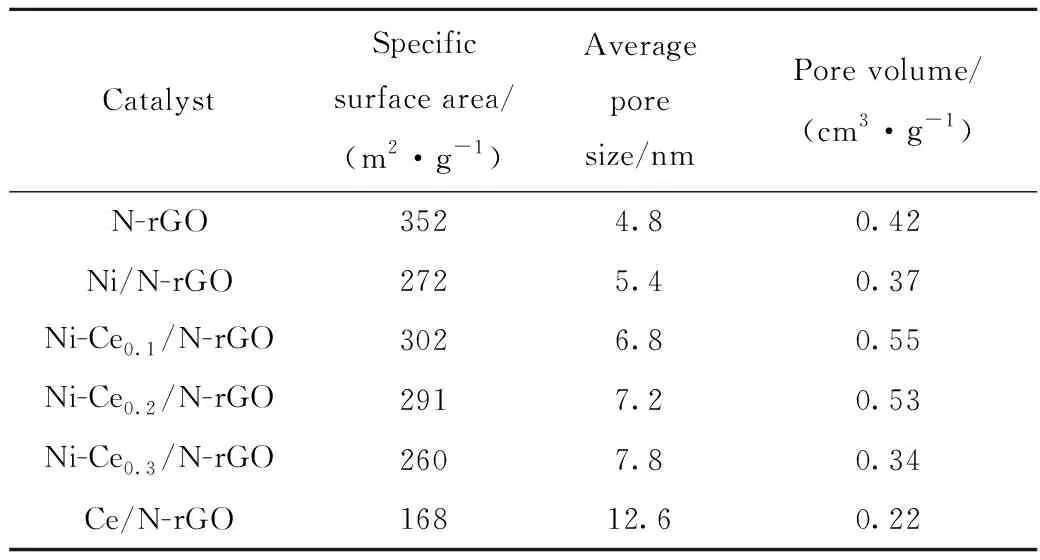
表1 N-rGO載體和Ni-Cen/N-rGO催化劑樣品的物理結構性質Table 1 Physical structural properties of N-rGO carriers and Ni-Cen/N-rGO catalyst samples
由表1可知:載體N-rGO負載金屬后比表面積減小,這是因為引入金屬占據了載體的表面,從而導致催化劑的比表面積減小。而在Ni/N-rGO中加入適當的稀土金屬Ce后,催化劑的比表面積、孔徑和孔體積顯著增大,說明適當Ce的加入使Ni分散更均勻。隨著n(Ce)/n(Ni)的增大,催化劑的比表面積和孔體積先增大后減小,可能是因為組分Ce對N-rGO的層狀結構有支撐作用,使孔體積增大,而當Ce添加過多,氧化鈰會覆蓋催化劑表面使孔體積降低。因此加入適當的Ce有利于提高催化劑的物性。
圖1為Ni-Cen/N-rGO催化劑樣品的N2吸附-脫附等溫曲線。由圖1可知:Ni/N-rGO和Ni-Cen/N-rGO催化劑的N2吸附-脫附等溫線都為Ⅳ型等溫線,這表明催化劑的孔結構都為介孔結構。催化劑的滯后環相對壓力(p/p0)都在0.4~1.0區間之內,其中Ni-Ce0.3/N-rGO和Ce/N-rGO是H4型遲滯回線,孔徑為微孔和介孔混合孔;其他3種催化劑是H2型遲滯回線,孔徑為介孔結構。因此,Ce的引入對催化劑孔結構有較大影響。
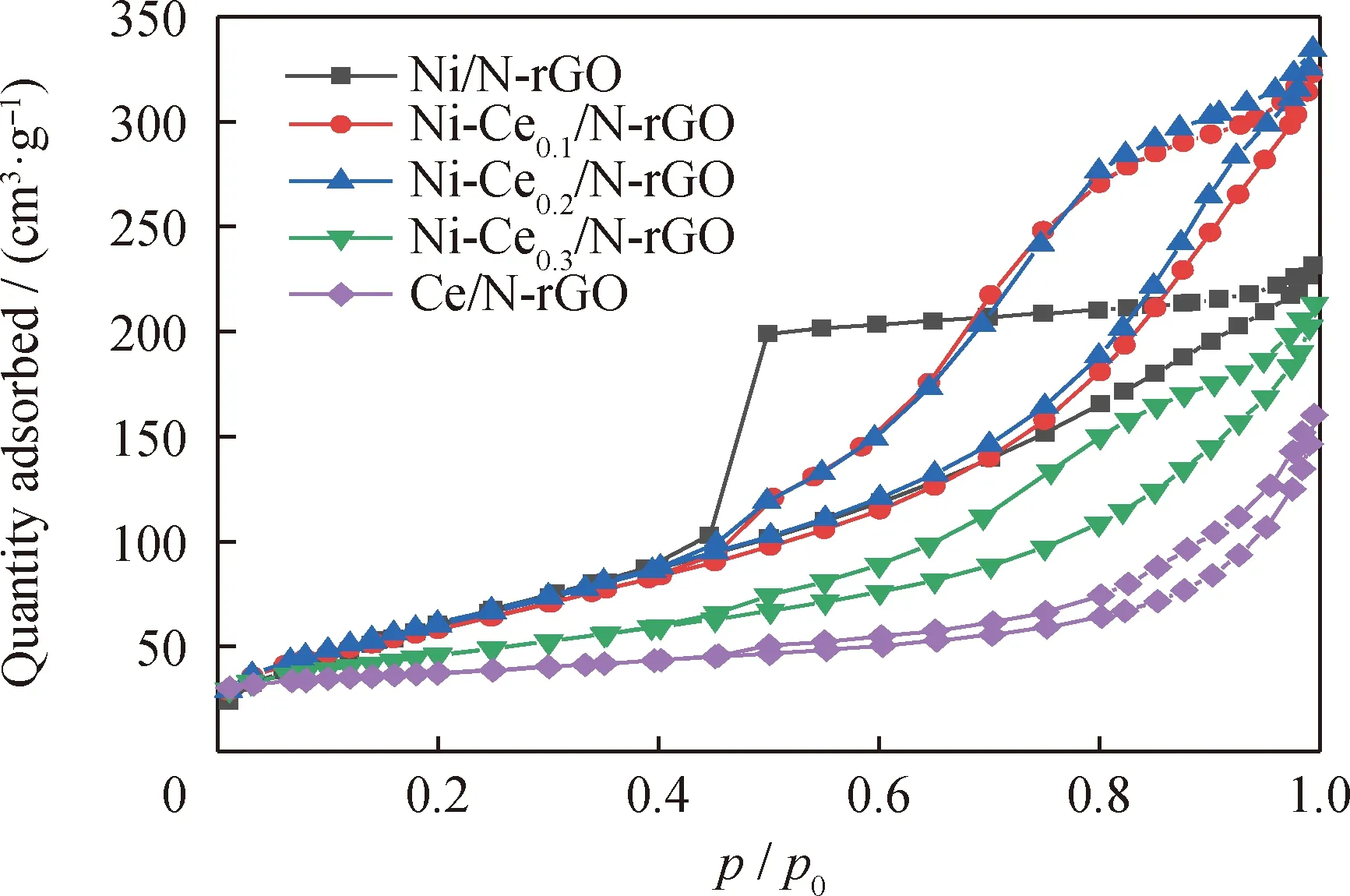
圖1 Ni-Cen/N-rGO催化劑樣品的N2吸附-脫附等溫曲線Fig.1 N2 adsorption-desorption isothermal curves of Ni-Cen/N-RGO catalyst samples
2.1.2 XRD表征
Ni-Cen/N-rGO催化劑樣品的XRD譜圖,如圖2所示。
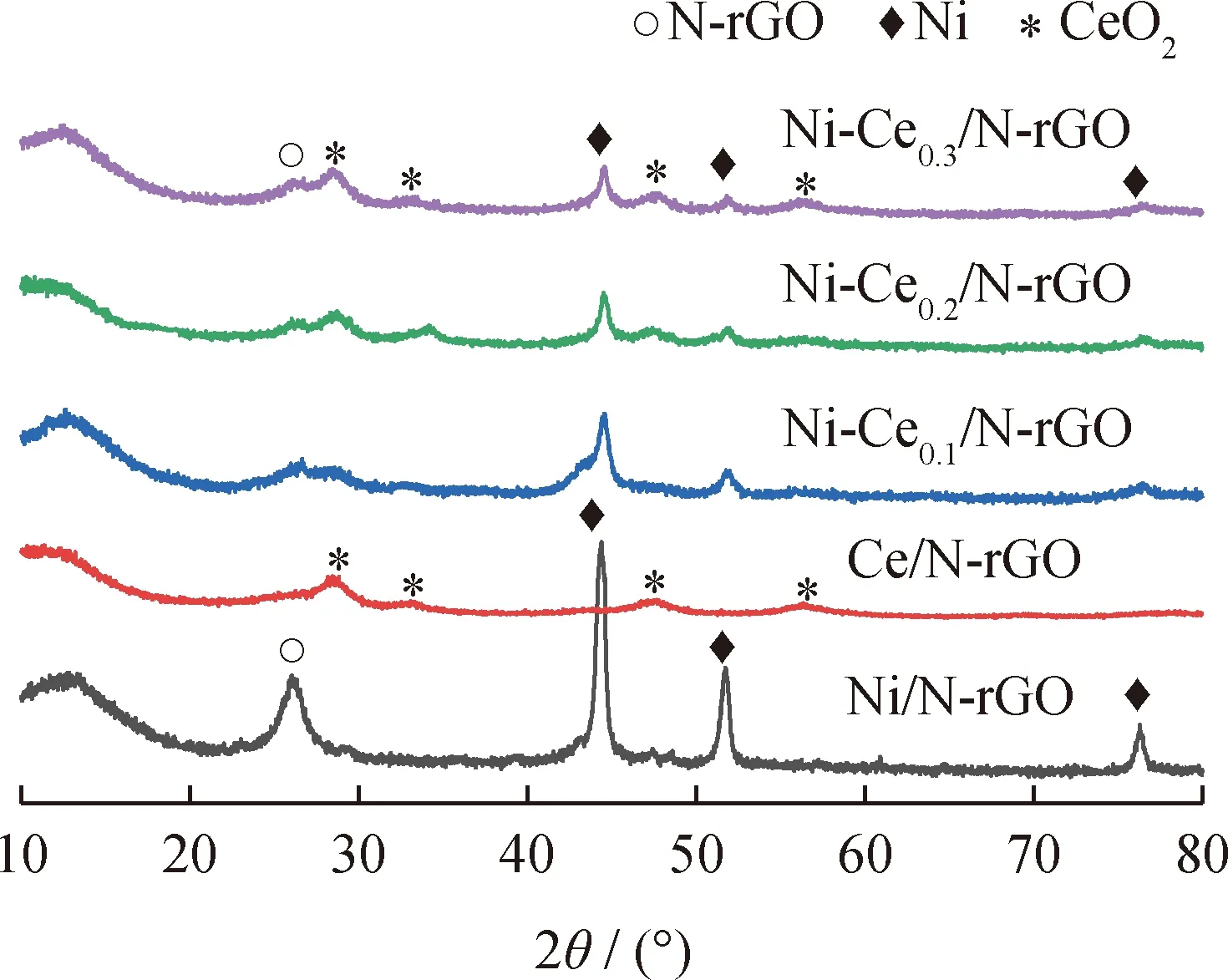
圖2 Ni-Cen/N-rGO催化劑樣品的XRD譜圖Fig.2 XRD patterns of Ni-Cen/N-rGO catalyst samples
由圖2可知:2θ在26.3°處對應N-rGO的(002)特征衍射峰;2θ在44.5°、51.7°和76.3°處對應金屬Ni的(111)、(200)、(220)特征衍射峰(JCPDS No. 04-0850)[18];2θ在28.6°、33.0°、47.6°和56.3°處對應二氧化鈰的(111)、(200),(220)、(311)面心立方螢石結構(JCPDS No.34-0394)特征衍射峰,說明Ce成功添加在Ni/N-rGO催化劑中[19]。相比于Ni/N-rGO催化劑尖銳的衍射峰,隨著Ce含量的增加,金屬Ni的衍射峰強度在逐漸變弱,峰形變寬,這表明金屬Ni顆粒變小。因此加入適量的Ce不僅會促使金屬Ni均勻分散,還能抑制金屬Ni顆粒團聚,但當加入的Ce過多時會覆蓋活性金屬Ni,因此Ce的添加量應適宜。
2.1.3 FT-IR表征
圖3為Ni-Cen/N-rGO催化劑樣品的FT-IR譜圖。由圖3可知,5種催化劑的紅外光譜圖相似,在3462 cm-1處對應O—H的伸縮振動峰,1620 cm-1處對應C=C的伸縮振動峰,1370 cm-1處對應O—H的變形振動峰,1070、1026 cm-1處對應C—O的伸縮振動峰,說明催化劑中含有石墨烯的基本官能團。1601 cm-1對應C=N的伸縮振動峰,1147 cm-1對應C—N的伸縮振動峰,說明催化劑中氮原子都成功摻雜。與N-rGO相比,隨著金屬Ni的負載石墨烯的含氧官能團吸收強度變小,原因可能是Ni分散石墨烯載體表面占據了其部分含氧官能團;且加入組分Ce后石墨烯含氧官能團的吸收強度變弱更明顯,這說明堿性組分Ce的添加有利于減弱石墨烯表面含氧官能團的酸性質。
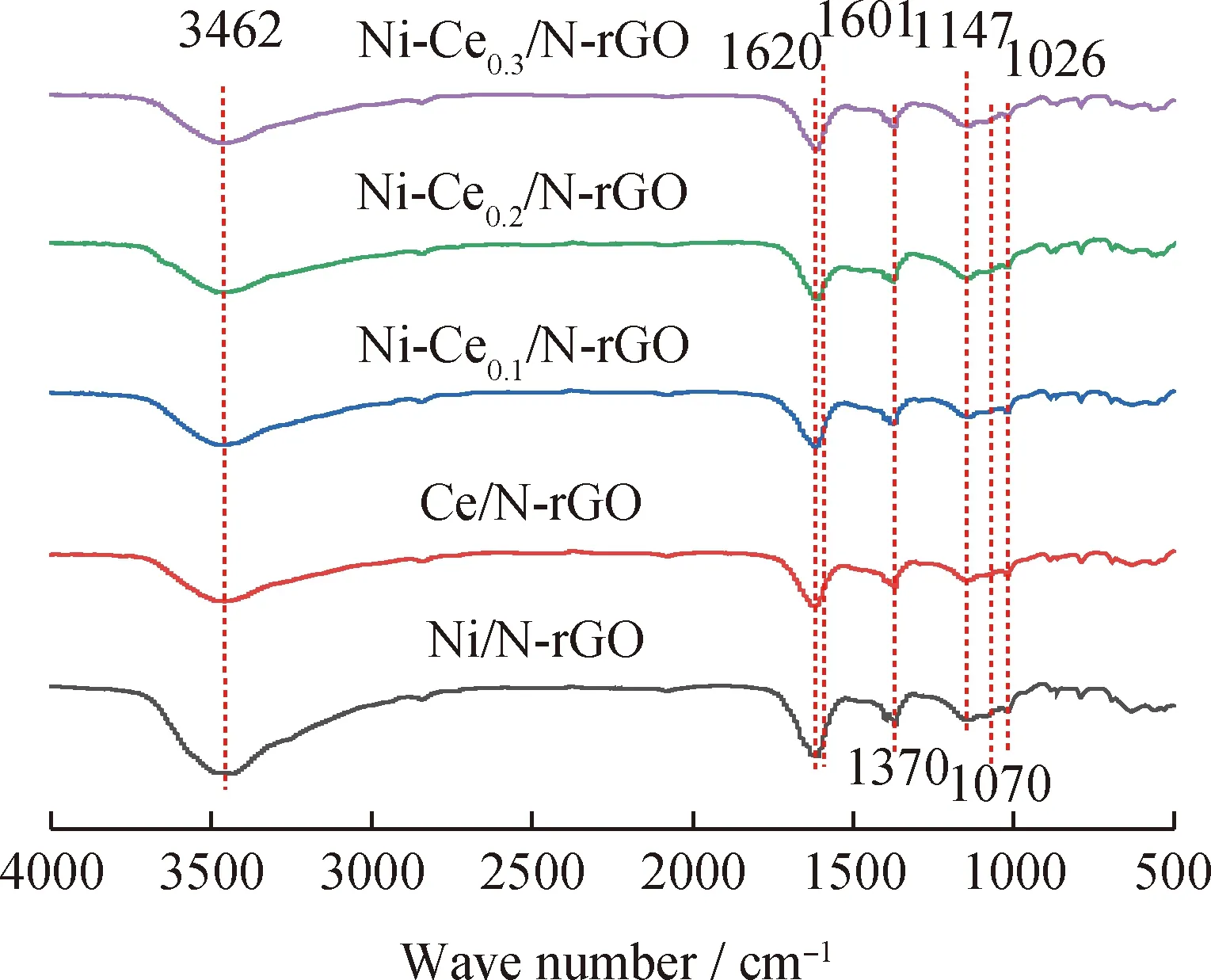
圖3 Ni-Cen/N-rGO催化劑樣品的FT-IR譜圖Fig.3 FT-IR spectra of Ni-Cen/N-rGO catalyst samples
2.1.4 SEM和EDS表征
圖4為N-rGO載體和Ni-Cen/N-rGO催化劑樣品的SEM照片。

圖4 N-rGO載體和Ni-Cen/N-rGO催化劑樣品的SEM照片Fig.4 SEM images of N-rGO carriers and Ni-Cen/N-rGO catalyst samples(a) N-rGO; (b) Ni/N-rGO; (c) Ni-Ce0.2/N-rGO; (d) Ni-Ce0.3/N-rGO
由圖4可知:N-rGO載體表面透明、無序、有薄紗褶皺且呈大片層二維疊狀結構;Ni-Cen/N-rGO催化劑的載體表面都有顆粒狀物質附著,其中Ni/N-rGO催化劑分散均勻但表面出現大顆粒團聚現象,Ni-Ce0.2/N-rGO催化劑形貌有明顯變化,表面顆粒變小分散更均勻,Ni-Ce0.3/N-rGO催化劑表面顆粒又出現了明顯的團聚。這說明Ni和Ce成功負載,且在加入適當組分Ce后可有效改善金屬Ni在載體表面的團聚現象;但加入的Ce過多時,二氧化鈰會占據催化劑的活性位點堵塞孔道,從而影響催化劑的催化性能。
表2為Ni-Ce0.2/N-rGO催化劑樣品中各元素的含量。由表2可知:C、N元素的質量分數分別為42.09%、3.49%,說明載體為N-rGO;Ce元素和Ni元素質量比接近0.2,催化劑中還有較多O元素,可能來源于N-rGO所攜帶的含氧基團和氧化鈰中的氧。
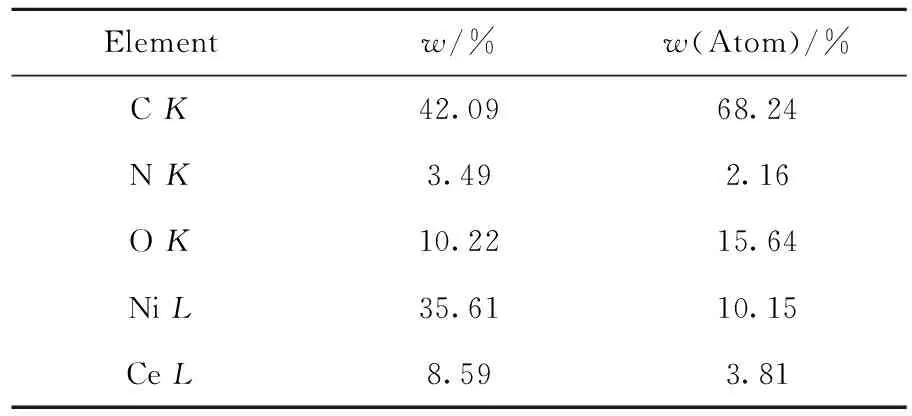
表2 Ni-Ce0.2/N-rGO催化劑樣品中各元素的質量分數Table 2 Mass fraction of each element in the Ni-Ce0.2/N-rGO catalyst samples
2.2 Ni/N-rGO催化劑反應性能評價結果
2.2.1 不同金屬組分對反應性能的影響
在反應溫度150 ℃、反應壓力0.4 MPa、反應時間2.0 h、催化劑用量0.2 g的條件下,考察不同金屬組分改性的Ni/N-rGO催化劑作用下苯酚加氫制備環己酮反應的苯酚轉化率和環己酮選擇性,結果如圖5所示。
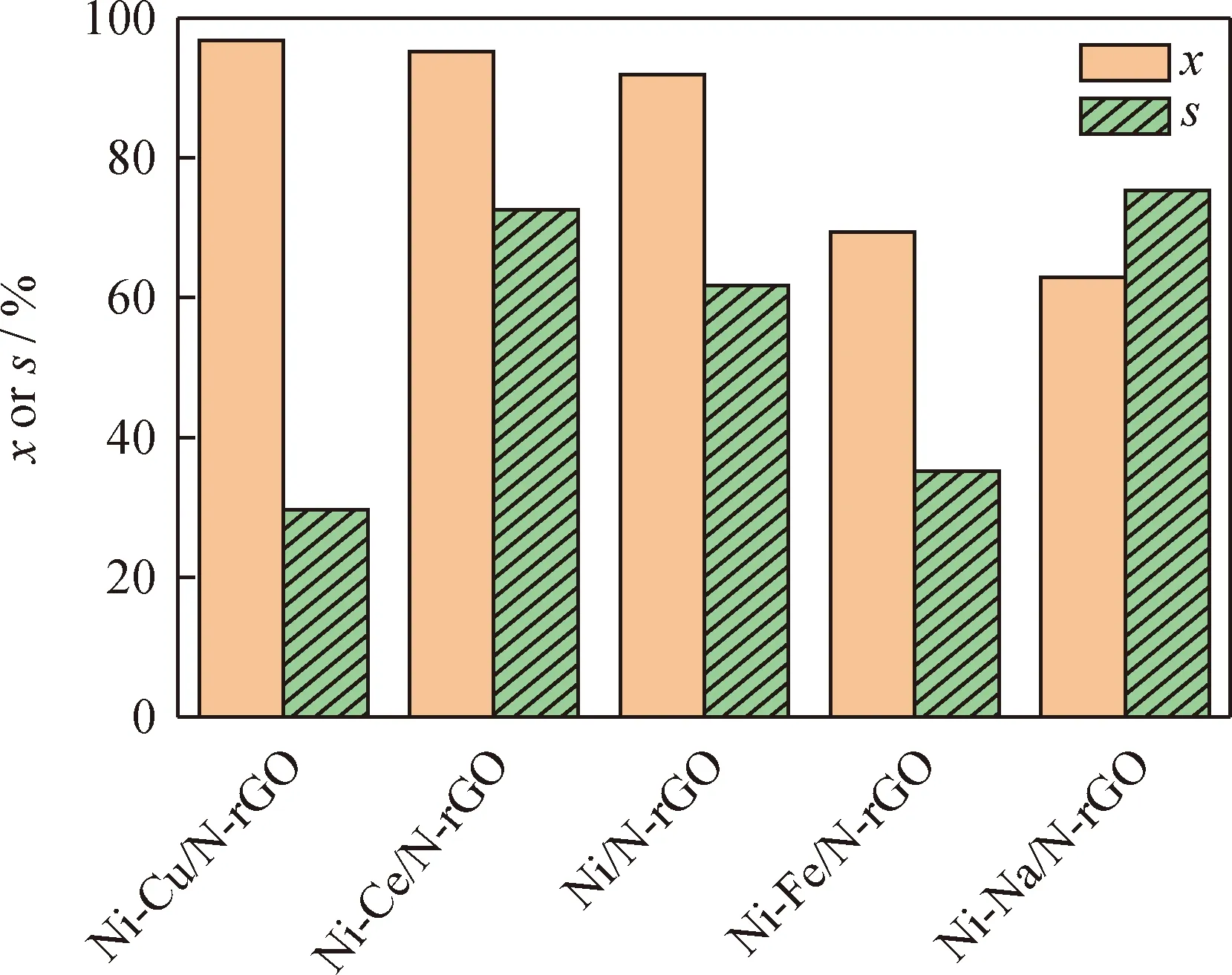
圖5 不同金屬組分改性的Ni/N-rGO催化劑作用下的苯酚轉化率(x)和環己酮選擇性(s)Fig.5 Phenol conversion (x) and and cyclohexanone selectivity (s) under the action of Ni/N-rGO catalysts modified by different metal componentsConditions: T=150 ℃; p=0.4 MPa; t=2.0 h; m(Catalyst)=0.2 g
由圖5可知:在反應條件相同的情況下,與金屬改性前催化劑的作用效果相比,Ni/N-rGO催化劑中加入Ce和Cu改性后,催化劑的反應活性有所增加,加入Fe和Na改性后催化劑的反應活性明顯降低;而加入Ce和Na改性后的催化劑作用下環己酮的選擇性會提高[15]。原因可能有:一是Ce的加入使金屬Ni在載體表面均勻分散,從而提高了Ni基催化劑的活性和選擇性;二是Ce和Na的堿性性質有利于提高環己酮選擇性;三是Cu的富電子特性和金屬Ni有較好的相互作用,但二者間過多的電子轉移促進了環己酮進一步反應生成副產物。所以加入組分Ce有利于提高催化劑的活性和選擇性。
2.2.2 Ce含量對反應性能的影響
在反應溫度150 ℃、反應壓力0.4 MPa、反應時間2.0 h、催化劑用量0.2 g的條件下,考察Ce含量對Ni/N-rGO催化劑作用下的苯酚轉化率和環己酮選擇性的影響,結果如表3所示。
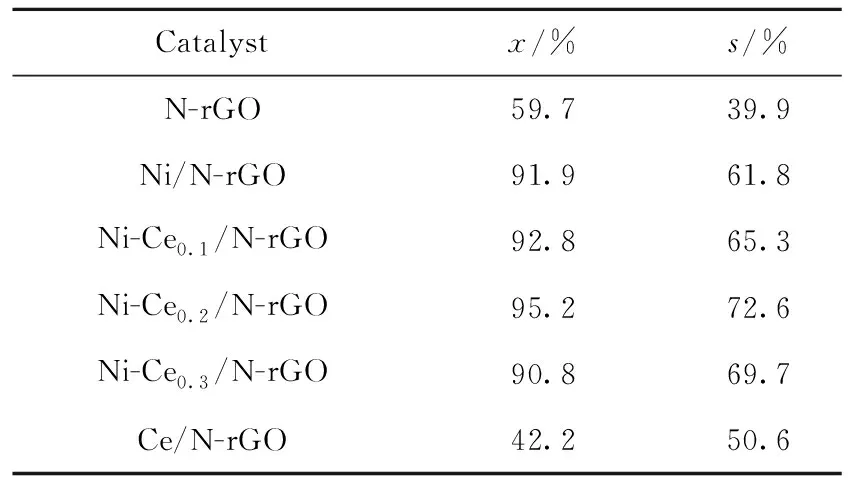
表3 Ce含量對Ni/N-rGO催化劑作用下苯酚轉化率(x)和環己酮選擇性(s)的影響Table 3 The effect of Ce content on phenol conversion (x) and cyclohexanone selectivity (s) under the action of Ni/N-rGO catalysts
由表3可知:隨著n(Ce)/n(Ni)的增大,催化劑的活性和環己酮選擇性先增大后減小;當n(Ce)/n(Ni)為0.2時,催化劑的活性和環己酮選擇性最大,苯酚轉化率達到95.2%,環己酮選擇性達到72.6%。原因可能有2點:一是加入Ce使Ni在載體N-rGO表面分散均勻;二是加入堿金屬Ce減弱了石墨烯表面含氧官能團的酸性性質,從而提高了催化劑的活性及環己酮選擇性,但是當Ce過量后,易形成大顆粒占據催化劑的活性位點。因此,在Ni/N-rGO加入適量Ce不僅可以提高苯酚轉化率還能提高環己酮選擇性。
2.3 Ni-Ce/N-rGO催化劑反應機理
根據理論和表征結果發現,Ce作為助劑可以提高Ni在載體表面的分散性,暴露更多Ni的活性位點,從而提高催化活性。Ni-Ce/N-rGO催化苯酚可能的反應機理如圖6所示。氮摻雜可增強石墨烯與苯酚的相互作用,苯酚通過羥基與N-rGO可能會形成O—H…N,有利于環己酮的生成[20];具有富電子性能的N原子可促進活性金屬Ni對H2吸附解離得到活潑氫,活化后的氫使苯環部分氫化生成環己烯醇,然后迅速異構化生成環己酮,提高了苯酚的轉化率[21];組分Ce的添加增加了催化劑的堿性位點,苯酚以非平面方式吸附在載體表面,抑制其進一步加氫生成環己醇,有利于提高環己酮的選擇性。
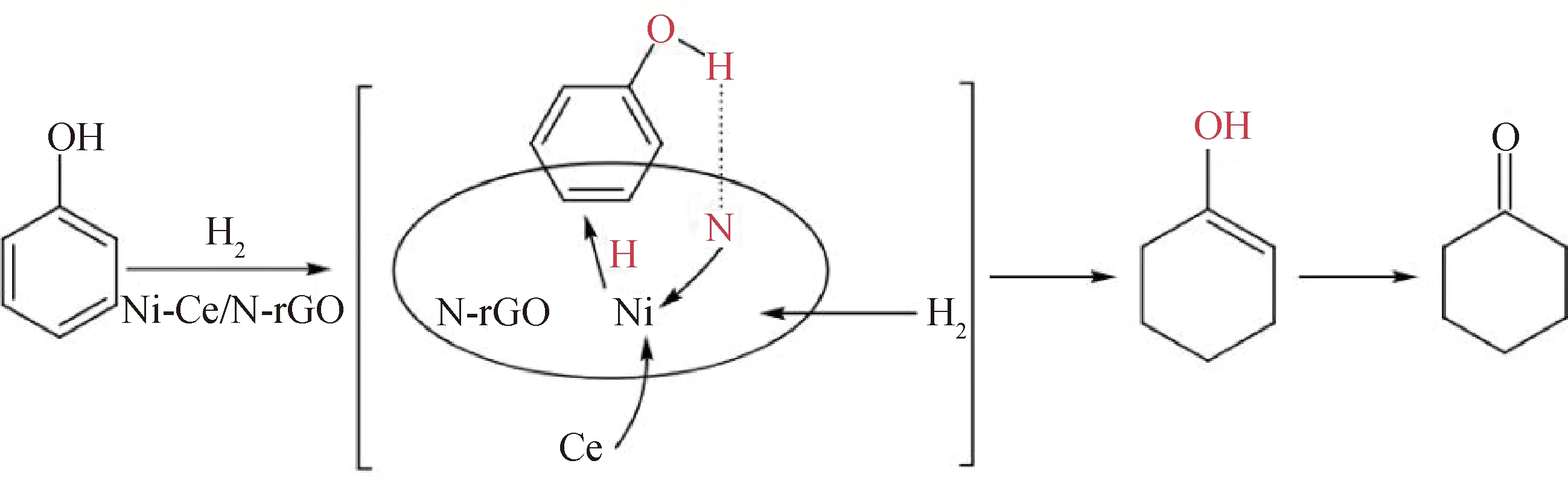
圖6 Ni-Ce/N-rGO催化劑催化苯酚加氫制環己酮反應機理Fig.6 Reaction mechanism for hydrogenation of phenol to cyclohexanone catalyzed by Ni-Ce/N-rGO catalyst
3 結 論
(1)對Ni-Ce/N-rGO催化劑進行表征,發現催化劑表面呈多孔結構,比表面積和孔徑比Ni/N-rGO催化劑明顯增大,表面團聚的Ni大顆粒基本消失,說明組分Ce的添加促使Ni在載體表面分散更均勻,提高了催化劑的催化性能。
(2)在反應溫度150 ℃、反應壓力0.4 MPa、反應時間2.0 h、催化劑用量0.2 g的條件下,經過比較不同金屬組分改性的Ni/N-rGO催化劑催化苯酚加氫反應性能,發現添加金屬組分Ce的催化劑反應效果最優。當n(Ce)/n(Ni)為0.2時制備的Ni-Ce/N-rGO催化劑作用下苯酚轉化率為95.2%,環己酮選擇性為72.6%。
(3)Ni基催化劑分散性較差、易發生團聚、成本低,加入稀土類金屬組分Ce后雖能得到一定改善,但是與貴金屬比較催化效果還不夠好,需繼續研究改善。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
