Nec-1和Z-VAD-FMK對正畸牙移動中caspase8、MLKL和RANKL表達的影響
2023-02-12 12:08:08張嚴勻張苗苗
醫學研究雜志 2023年1期
關鍵詞:實驗
李 易 王 賀 張 曼 張嚴勻 張苗苗
正畸牙移動(orthodontic tooth movement, OTM)必然伴隨著牙周組織的改建,OTM中壓力側細胞的死亡形式以及對牙周組織改建的影響機制是當前研究的熱點。凋亡和壞死性凋亡均屬于基因控制下的程序性細胞死亡,caspase8是外源性凋亡途徑的起始caspase,當其被抑制或敲除,將激活壞死性凋亡途徑,混合譜系激酶結構域樣蛋白(mixed lineage kinase domain-like protein, MLKL)則是破壞細胞質膜完整性、執行壞死性凋亡的效應蛋白[1]。壓力側牙周組織改建主要表現為破骨細胞性骨吸收,現已發現多種與破骨細胞形成相關的信號轉導機制,其中核因子κB受體活化因子配體(receptor activator of nuclear factor κB ligand, RANKL)信號通路在破骨細胞的分化誘導、激活和存活中發揮了關鍵作用[2]。
近年來研究表明,OTM中壓力側的牙周組織細胞可發生凋亡,并與破骨細胞的分化、募集有關[3,4]。課題組前期實驗首次在壓力側牙周組織中發現部分細胞死亡是由壞死性凋亡介導[5]。雖然目前已對凋亡和壞死性凋亡信號通路做了深入研究,但是關于兩者在OTM中的相互聯系及對牙周組織改建的影響機制尚不完全清楚。本實驗通過建立大鼠OTM模型,分別使用Nec-1和Z-VAD-FMK后觀察caspase8、MLKL和RANKL在壓力側牙周組織中的變化,初步探討壓力側牙周組織凋亡和壞死性凋亡的相互聯系及對RANKL表達的影響。
材料與方法
1.主要材料與試劑:鎳鈦螺旋拉簧購自北京有研億金新材料有限公司,粘接劑、流動樹脂購自美國3M公司,正畸測力計購自杭州西湖生物材料有限公司,Nec-1、Z-VAD-FMK購自美國MedChemExpress公司,即用型正常山羊血清、DAB顯色試劑盒、caspase8、RANKL抗體購自武漢博士德生物工程有限公司,MLKL抗體購自美國Affinity公司,兔二步法試劑盒購自北京中杉金橋生物技術有限公司。
2.實驗動物及分組:80只7周齡健康雄性SD大鼠,體質量為200±10g,購自遼寧長生生物技術股份有限公司[實驗動物許可證號:SCXK(遼)2020-0001,倫理審批號:2021084]。適應性喂養1周,采用隨機數字表法分為對照組(C組)、加力組(F組)、加力+Nec-1組(FN組)、加力+Z-VAD-FMK組(FZ組),每組各20只。
3.動物模型的建立:10%的水合氯醛以0.3ml/100g的劑量腹腔注射麻醉大鼠,將其仰臥固定在實驗臺上,在上頜左側第一磨牙和2顆上切牙之間用結扎絲固定1段鎳鈦拉簧,力量50g,建立以上頜2顆切牙為支抗拉左側第一磨牙近中移動模型。C組無加力裝置,F、FN、FZ組建立OTM模型,C組、F組于加力前半小時用微量進樣器在大鼠上頜左側第一磨牙頰腭側牙齦黏膜局部注射50μl 0.9% NaCl溶液,FN組、FZ組以同樣方式分別注射等體積1mmol/L的Nec-1和0.25mmol/L的Z-VAD-FMK,每隔2天注射1次。每天檢查1次加力裝置是否穩固,若有松動或脫落及時重新安置。
4.切片制備:獲取上頜磨牙及牙槽骨的組織塊,4%多聚甲醛溶液外固定48h,15%EDTA液(pH值為7.2)脫鈣4周,脫水、透明、浸蠟、包埋、切片(厚5μm),光鏡下挑選完整的切片用于蘇木精-伊紅(hematoxylin-eosin, HE)染色及免疫組化染色。
5.免疫組化染色及圖像分析:烤片后進行脫蠟水化,抗原修復,血清封閉,分別滴加MLKL、caspase8、RANKL多克隆抗體4℃過夜,二抗37℃孵育20min,光鏡下DAB顯色觀察,自來水沖洗終止,蘇木精復染,氨水返藍,脫水透明,封片。光鏡下觀察并截取圖像,采用Image-Pro Plus 6.0軟件對圖像進行平均吸光度值測量。設定第1、3、5、7、14天5個時間點,觀察每組大鼠對應的形態學改變,以及caspase8、MLKL、RANKL的免疫組化結果。
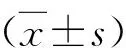
結 果
1.壓力側牙周組織的形態學改變:HE染色可見C組第一磨牙遠中根壓力側牙周膜成纖維細胞排列規則,未見骨吸收陷窩。加力第1天時,牙周膜成纖維細胞排列紊亂;第3天時,細胞數目明顯減少,形成玻璃樣變;第5天起,出現破骨細胞及骨吸收陷窩,玻璃樣變范圍有所減少;第7天時,玻璃樣變基本清除;第14天時,成纖維細胞排列規則,未見破骨細胞及骨吸收陷窩(圖1)。

圖1 壓力側牙周組織的變化(HE染色,×200)A.C組第7天;B.F組第1天;C.F組第3天;D.F組第5天;E.F組第7天;F.F組第14天。R.牙根;PDL.牙周膜;AB.牙槽骨;白色箭頭示玻璃樣變;黑色箭頭示破骨細胞
2.caspase8的免疫組化結果:C組各時間點caspase8的表達比較差異無統計學意義(P>0.05)。F、FN、FZ組第1、3、5、7天的表達均高于C組(P<0.05)。F組中caspase8的表達隨加力時間逐漸增加,第3天達到高峰,之后減少。FN組各時間點的表達與相應F組中的表達比較,差異均無統計學意義(P>0.05)。與F組和FN組比較,FZ組各時間點的表達明顯降低(P<0.05),詳見圖2、表1。
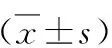
表1 各組大鼠不同時間點第一磨牙壓力側牙周組織caspase8表達的平均吸光度值

圖2 各組大鼠第3天時caspase8的表達情況(免疫組化染色,×200)A.C組第3天;B.F組第3天;C.FN組第3天;D.FZ組第3天。R.牙根;PDL.牙周膜;AB.牙槽骨
3.MLKL的免疫組化結果:C組各時間點MLKL的表達比較,差異無統計學意義(P>0.05)。F組第1、3、5天的表達均較C組增強,加力第1天時即達到表達高峰,隨著加力時間延長呈降低趨勢。FN組第1、3天的表達較C組增強;第1、3、5天的表達與相應F組中的表達比較顯著降低(P<0.05)。FZ組第1、3、5天的表達均較C、F、FN組增強(P<0.05)。第7、14天各組MLKL的表達比較,差異均無統計學意義(P>0.05),詳見圖3、表2。
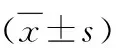
表2 各組大鼠不同時間點第一磨牙壓力側牙周組織MLKL表達的平均吸光度值

圖3 各組大鼠第1天時MLKL的表達情況(免疫組化染色,×200)A.C組第1天;B.F組第1天;C.FN組第1天;D.FZ組第1天;R.牙根;PDL.牙周膜;AB.牙槽骨
4.RANKL的免疫組化結果:C組各時間點RANKL的表達比較,差異無統計學意義(P>0.05)。F組RANKL表達隨加力時間逐漸增加,第5天達到高峰,之后減少,各時間點的表達均高于C組(P<0.05)。FN組第1、3、5、7天的表達較F組減少,但較C組增多(P<0.05)。FZ組第3、5、7天的表達較F、FN組減少(P<0.05)。第14天時,F、FN、FZ組RANKL的表達比較,差異無統計學意義(P>0.05),但均高于C組(P<0.05),詳見圖4、表3。

圖4 各組大鼠第5天時RANKL的表達情況(免疫組化染色,×200)A.C組第5天;B.F組第5天;C.FN組第5天;D.FZ組第5天。R.牙根;PDL.牙周膜;AB.牙槽骨
表3 各組大鼠不同時間點第一磨牙壓力側牙周組織RANKL表達的平均吸光度值
討 論
凋亡介導的細胞死亡與caspase家族活化密切相關,caspase8是外源性凋亡途徑的啟動caspase,進而激活下游caspase3執行凋亡活動。MLKL是壞死性凋亡發生的關鍵,磷酸化的MLKL發生寡聚并轉移至質膜,破壞質膜完整性導致細胞壞死[6]。本實驗觀察到牙周組織在加力第1天時即出現明顯的caspase8表達,第3天時達到表達高峰,MLKL同樣在加力第1天時出現明顯表達,之后便呈降低趨勢,提示牙周組織細胞凋亡和壞死性凋亡主要發生在正畸加力初期。同時,caspase8的表達也說明了壓力側牙周組織凋亡可以通過外源性凋亡途徑激活。但一些研究觀察到caspase3在牙齒移動7天后仍有增加,由于caspase3是多條凋亡途徑下游共同的效應蛋白酶,因此細胞凋亡可能存在于整個OTM過程中且涉及多條凋亡信號通路,而外源性凋亡途徑可能主要介導了牙齒移動早期的凋亡發生[7]。
研究表明,Z-VAD-FMK在抑制caspase8活性阻斷凋亡發生的同時可以增強壞死性凋亡[8]。本實驗使用Z-VAD-FMK后,caspase8表達早期明顯降低,MLKL表達明顯升高,這一結果與上述研究一致。Nec-1通過變構抑制受體相互作用蛋白1(receptor-interacting protein1, RIP1)激酶活性從而有效抑制壞死性凋亡,但是否影響凋亡目前的研究仍有爭議[9]。本實驗使用Nec-1后,MLKL水平明顯降低,caspase8水平無明顯變化,說明Nec-1可以抑制MLKL的表達而對caspase8的表達無影響,但由于凋亡信號通路復雜,且本實驗檢測指標有限,僅caspase8的表達不足以完全反映凋亡水平,因此Nec-1對OTM牙周組織細胞凋亡的影響仍需進一步研究。
RANKL是一種來自腫瘤壞死因子(tumour necrosis factor, TNF)家族的膜相關細胞因子,在牙周組織中可以由成骨細胞、成纖維細胞、骨細胞以及成牙骨質細胞分泌,與破骨前體細胞表面的核因子κB受體活化因子(receptor activator of nuclear factor κB, RANK)結合,通過激活核因子κB(nuclear factor kappa B, NF-κB)和絲裂原活化蛋白激酶(mitogen activated protein kinase, MAPK)等下游信號通路誘導破骨細胞分化成熟[10~14]。研究表明,壓應力可以上調牙周組織中RANKL的表達水平,并與caspase3的表達呈正相關[4]。Kaplan等[3]利用雙磷酸鹽類似物IG9402抑制骨細胞凋亡發現會阻礙破骨細胞和巨噬細胞的形成,認為牙周膜細胞凋亡和骨細胞凋亡在OTM骨吸收中發揮了同等重要的作用。本實驗正畸加力后觀察到RANKL表達以時間依賴性的方式增加,加力第5天時達到表達高峰,隨后逐漸減少。使用凋亡抑制劑Z-VAD-FMK后,第1、3、5、7天的RANKL表達較加力組顯著減少,提示牙周組織細胞凋亡可以上調RANKL表達以促進破骨細胞生成。
不同于凋亡,壞死性凋亡使質膜破裂導致細胞內容物釋放進而啟動非感染性炎性反應,此外RIP1和RIP3也可直接產生炎性細胞因子[15]。本實驗使用壞死性凋亡抑制劑Nec-1后,第1、3、5、7天的RANKL表達較加力組明顯減少,說明壞死性凋亡參與了壓力側牙周組織上調RANKL的過程,這可能與壞死性凋亡介導的炎性反應有關。據報道,炎性細胞因子如TNF-α,既可以刺激間充質細胞分泌RANKL,又可以協同RANKL作用于RANK,增強破骨細胞生成的信號轉導[16,17]。抑制壞死性凋亡可以通過減輕炎性反應進而削弱破骨細胞的形成能力。然而,壞死性凋亡對破骨細胞的調控作用似乎是有限的,因為FZ組盡管增強了壞死性凋亡,但RANKL表達均較F組和FN組減少,可能是由于抑制凋亡后牙周組織發生過多壞死,致使分泌RANKL的細胞來源減少。這也進一步說明了破骨細胞分化必須存在RANKL和RANK,雖然壞死性凋亡引起了炎性反應增加,但是在缺乏RANKL和RANK的條件下炎性細胞因子無法獨立誘導破骨細胞形成[17]。
綜上所述,OTM中壓力側牙周組織可同時發生凋亡和壞死性凋亡,抑制凋亡可以促進壞死性凋亡。此外,兩者均可引起壓力側牙周組織RANKL高表達,參與牙周組織改建,為進一步研究正畸力作用下細胞死亡對牙周組織改建的影響機制奠定了基礎。
猜你喜歡
作文·小學低年級(2025年2期)2025-02-13 00:00:00
小雪花·小學生快樂作文(2024年11期)2024-12-31 00:00:00
作文·小學低年級(2024年2期)2024-04-29 00:00:00
作文·小學低年級(2023年3期)2023-04-29 00:00:00
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
小主人報(2022年4期)2022-08-09 08:52:06
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
