含氮芥和蒽環的新型α,β-不飽和酮的合成、表征與抗腫瘤活性
2023-03-03 08:08:26黃偉婷何露欣李世曉丘文樺王戴妮龔曉璇梁遠維
合成化學 2023年2期
黃偉婷, 何露欣, 李世曉, 丘文樺, 趙 曦, 王戴妮, 龔曉璇, 梁遠維*
(1. 廣東海洋大學 化學與環境學院,廣東 湛江 524088; 2. 遼寧省環保集團科源環境技術有限公司,遼寧 沈陽 110161)
蒽類化合物是天然產物中一類比較重要的活性成分,主要分布在蓼科、鼠李科、茜草科、豆科、百合科和玄參科等高等植物中。蒽類化合物存在形式多樣,包括氧化蒽酚、蒽酚和蒽酮等[1]。許多蒽類衍生物具有不同的生理活性,主要表現為降血脂、利尿、抗氧化和抗腫瘤等,尤其是抗腫瘤方面表現出較大的潛力[2]。但由于蒽類衍生物種類繁多,活性參差不齊,很多情況下需通過原材料進行分離提純,大大限制了蒽類化合物的應用和開發。因此,對蒽類化合物進行結構修飾,獲得理想活性的藥物,具有重要的現實意義。
α,β-不飽和酮衍生物在天然產物和藥物分子中分布廣泛,由于其獨特的結構特征,在合成化學領域有重要研究前景[3]。α,β-不飽和酮不僅是重要的有機合成中間體,廣泛應用于化學和材料等領域,還具有顯著的生物藥理活性及獨特的可塑性結構,在抗腫瘤、抗炎、抗氧化和抗菌等方面有潛在應用價值[4]。α,β-不飽和酮衍生物在抗腫瘤方面具有細胞毒性小,選擇性高的特點[5]。
氮芥類藥物是最早應用于抗腫瘤作用的藥物之一,也是目前抗腫瘤藥用研發的重要方向之一。氮芥類藥物是一類高度活潑的基團,主要由氮芥部分和載體部分組成,在臨床上有廣泛的應用[6]。氮芥部分是抗腫瘤藥的活性功能基團,載體部分主要影響藥物的物化性質,以及在體內的吸收、分布等藥代動力學性質[7]。雖然氮芥類藥物抗瘤譜廣、對腫瘤細胞殺傷力強,但仍存在生物利用度低、選擇性差且毒副作用大等缺點[8]。
目前,關于含氮芥和蒽環的α,β-不飽和酮衍生物的抗腫瘤藥物尚未見報道,本文擬將氮芥、蒽環和α,β-不飽和酮等3個單元進行結合,合成含氮芥和蒽環的新型α,β-不飽和酮(1a和1b,圖1),其結構經紫外、熒光光譜、質譜和核磁共振等表征。通過MTT試驗法檢測目標化合物對A549、 786-O、 Hela和MDA-MB-231等4種腫瘤細胞的抗增殖活性,比較了2個化合物的活性,并進一步探討化合物對腫瘤細胞的抗轉移作用和對細胞周期的影響。
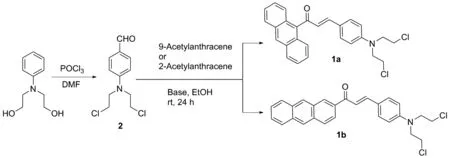
圖1 含氮芥和蒽環的α,β-不飽和酮的合成路線
1 實驗部分
1.1 儀器與試劑
BRUKER AVANCE 300/600 MHz型核磁共振儀(TMS為內標);Waters Xevo G2-XS Tof型飛行時間質譜儀;RF-6000型熒光分光光度計;TU-1901型紫外可見分光光度計;Thermo Multiskan FC型酶標儀;Beckman Gallios型流式細胞儀。
所用試劑均為分析純(畢得醫藥科技有限公司)。
1.2 1a和1b的合成通法
將N,N-二甲基甲酰胺(9 mmol, 657 mg),三氯氧磷(5 mmol, 767 mg)和N,N-二羥乙基苯胺(2 mmol, 362 mg)加入圓底燒瓶中,加熱至90 ℃,反應3 h。將液體倒入冰水中,用1M NaOH溶液調節pH≈7,過濾,濾餅依次用純水和冷乙醇洗滌兩次,真空干燥得中間體4-[雙-(2-氯乙基)氨基]苯甲醛(化合物2),淡黃色固體449 mg,產率 85%;1H NMR(300 MHz, DMSO-d6)δ: 9.72(s, 1H, CHO), 7.72(d,J=8.89 Hz, 2H, ArH), 6.90(d,J=8.89 Hz, 2H, ArH), 3.85(t,J=5.78Hz, 4H, CH2), 3.79(t,J=5.78 Hz, 4H, CH2); HR-MS(ESI)m/z: Calcd for C11H14Cl2NO{[M+H]+}246.0452, found 246.0468。
將化合物2(1 mmol, 246 mg)、 9-乙酰蒽或2-乙酰蒽(1 mmol,220 mg)和堿催化劑(NaOH, K2CO3或三乙胺,1 mmol)加入乙醇(13 mL)中,反應24 h。除去乙醇,殘余物經硅膠柱層析(洗脫劑:CH2Cl2∶CH3OH=45 ∶1,V∶V)純化得目標化合物1a和1b。
化合物1a:黃色固體368 mg,產率82%, m.p.127.6~128.6 ℃;1H NMR(600 MHz, DMSO-d6)δ: 8.73(s, 1H, ArH), 8.18(d,J=8.51Hz, 2H, ArH), 7.81(d,J=8.49 Hz, 2H, ArH), 7.57~7.52(m, 4H, ArH), 7.46(d,J=8.96 Hz, 2H, ArH), 7.22(d,J=16.07 Hz, 1H, CH), 7.05(d,J=16.07 Hz, 1H, CH), 6.73(d,J=8.96 Hz, 2H, ArH), 3.78(t,J=6.20 Hz, 4H, CH2), 3.72(t,J=6.20 Hz , 4H, CH2);13C NMR(150 MHz, DMSO-d6)δ: 198.96, 149.61, 148.52, 135.68, 131.58, 131.15, 129.15, 128.24, 128.06, 127.15, 126.09, 125.40, 124.88, 122.62 122.42, 52.12, 41.44; HR-MS(ESI)m/z: Calcd for C27H24Cl2NO{[M+H]+}448.1235, found 448.1234。
化合物1b: 黃色固體341 mg,產率76%, m.p.129.8~130.8 ℃;1H NMR(600 MHz, DMSO-d6)δ: 8.96(s, 1H, ArH), 8.72(s,1H, ArH), 8.31(d,J=8.70 Hz, 1H, ArH), 8.13(dd,J=8.17 Hz, 13.19 Hz, 2H, ArH), 7.94(d,J=6.80 Hz, 1H, ArH), 7.67(d,J=8.93 Hz, 2H, ArH), 7.64(dd,J=6.90 Hz, 8.62Hz, 1H, ArH), 7.62~7.56(m, 3H, ArH, CH), 7.39(d,J=15.61 Hz, 1H, CH), 6.94(d,J=8.97 Hz, 2H, ArH), 3.84(t,J=6.81 Hz, 4H, CH2), 3.78(t,J=6.11 Hz , 4H, CH2);13C NMR(150 MHz, DMSO-d6)δ: 194.49, 149.31, 146.34, 137.41, 132.22, 132.15, 131.89, 131.57, 131.46, 129.02, 128.33, 128.25, 127.81, 127.38, 126.65, 126.63, 124.91, 124.81, 123.26, 122.37, 112.44, 52.21, 41.51; HR-MS(ESI)m/z: Calcd for C27H24Cl2NO{[M+H]+}448.1235, found 448.1233。
1.3 體外抗增殖活性測試
將處于對數生長期的腫瘤細胞(A549、 786-O、 Hela和MDA-MB-231)接種于96孔板(接種密度2×103cells/well)。待細胞貼壁后,每孔加入不同濃度的1a或1b(0、 1、 2、 4、 8、 16、 32和64 μM),于37 ℃孵育72 h,吸棄96孔板內的培養基,每孔加入25 μL MTT試劑,在培養箱內孵育3 h。MTT在活細胞內被還原為不溶于水的藍紫色結晶甲臜(λmax=570 nm),而死細胞無此功能,因此甲臜形成的量與活細胞數成正比。用DMSO將其溶解后,通過酶標儀讀取每孔OD570數值,計算不同濃度藥物處理后細胞的存活率[9]。
1.4 細胞劃痕實驗
將對數生長期的Hela細胞接種于6孔板(4×105cells/well),待貼壁后,用槍頭在細胞生長的中央區域垂直劃線,碎片用PBS清洗并吸棄,添加培養基,加入不同濃度的化合物1a并孵育至設定的時間,吸棄培養基后加入Hoechst染料于37 ℃孵育染色30 min,用PBS清洗2次后,在倒置熒光顯微鏡下拍照[10]。
1.5 細胞周期實驗
將細胞接種于10 cm細胞培養皿中(1×106cells/well),貼壁后加入藥物,待藥物作用48 h后,將細胞消化收集于離心管,扣干,用PBS重懸1次再扣干,并用72%酒精固定溫度于4 ℃, 12 h后離心扣干,加入300 μL PI染色,避光孵育30 min。用尼龍網過濾后加入上機樣品管內,用流式細胞儀進行分析檢測,得出細胞處于G1期,S期和G2/M期的比例[11]。
2 結果與討論
2.1 合成分析
含氮芥和蒽環的α,β-不飽和酮的合成路線見圖1,起始原料為N,N-二羥乙基苯胺,第一步反應由POCl3和DMF組成的Vilsmeier-Haack試劑對苯環進行甲酰化反應[12],該反應為親電取代反應,過量的POCl3對2個羥基進行氯化反應得到化合物2。這里由于氮原子p軌道孤對電子與苯環形成p-π共軛,增大了苯環的電子云密度,活化了苯環,因此該甲酰化親電取代反應較為容易進行,產率也較高(85%)。第二步反應為典型的羥醛縮合反應[13],蒽環上的乙酰基先在堿的作用下生成碳負離子,接著進攻醛基的碳原子,發生親核加成反應,得到的β-羥基酮進一步脫水,生成α,β-不飽和酮,即為含氮芥和蒽環的α,β-不飽和酮1a和1b。反應溫度為室溫,反應時間為24 h。
羥醛縮合反應是制備α,β-不飽和醛酮的重要方法[14]。為了比較不同的催化劑對產率的影響,選擇了3種堿(NaOH、 K2CO3和三乙胺)作為催化劑進行篩選。其中強堿NaOH做催化劑時產率較高,1a和1b產率分別為82%和76%;而弱堿K2CO3催化時幾乎沒有產物生成,產率均小于1%;用有機堿三乙胺催化時產率也非常低,1a和1b產率僅有2%和3%,這是因為需要較強的堿才能將乙酰基的氫拔除,而K2CO3和三乙胺較難撥氫,碳負離子難以生成,無法得到較多的產物。
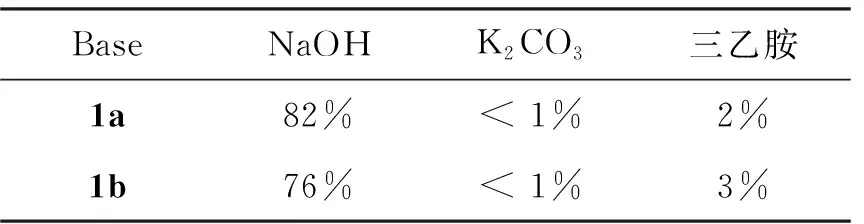
表1 不同堿催化劑對目標產物產率的影響
2.2 結構表征分析
氫譜在化合物的表征中是必不可少的,下面對化合物1a的核磁共振氫譜進行解析。圖2中δ8.73處為5號碳上的氫,單峰;δ8.18處為1號碳上的氫,受2號碳上的氫影響裂分為d峰,偶合常數為8.51 Hz;δ7.81處為4號碳上的氫,受3號碳影響裂分為d峰;δ7.57~7.52處為2號碳和3號碳上的氫(重疊);δ7.46處為8號碳上的氫受9號碳上的氫影響裂分為d峰,偶合常數為8.96 Hz;δ7.22處為7號碳上的氫裂分為d峰,偶合常數為16.07 Hz;δ7.05和6.74處分別為6號碳和9號碳上的氫,分別受7號碳和8號碳上的氫影響裂分為d峰;δ3.78和3.72處為10號和11號亞甲基的氫,各為三重峰。
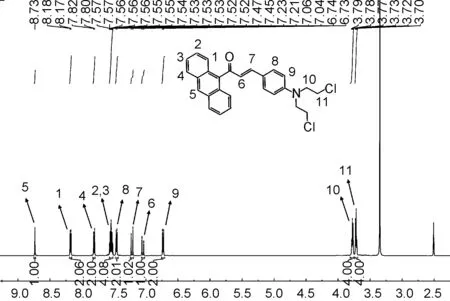
圖2 化合物1a的核磁共振氫譜
λ/nm
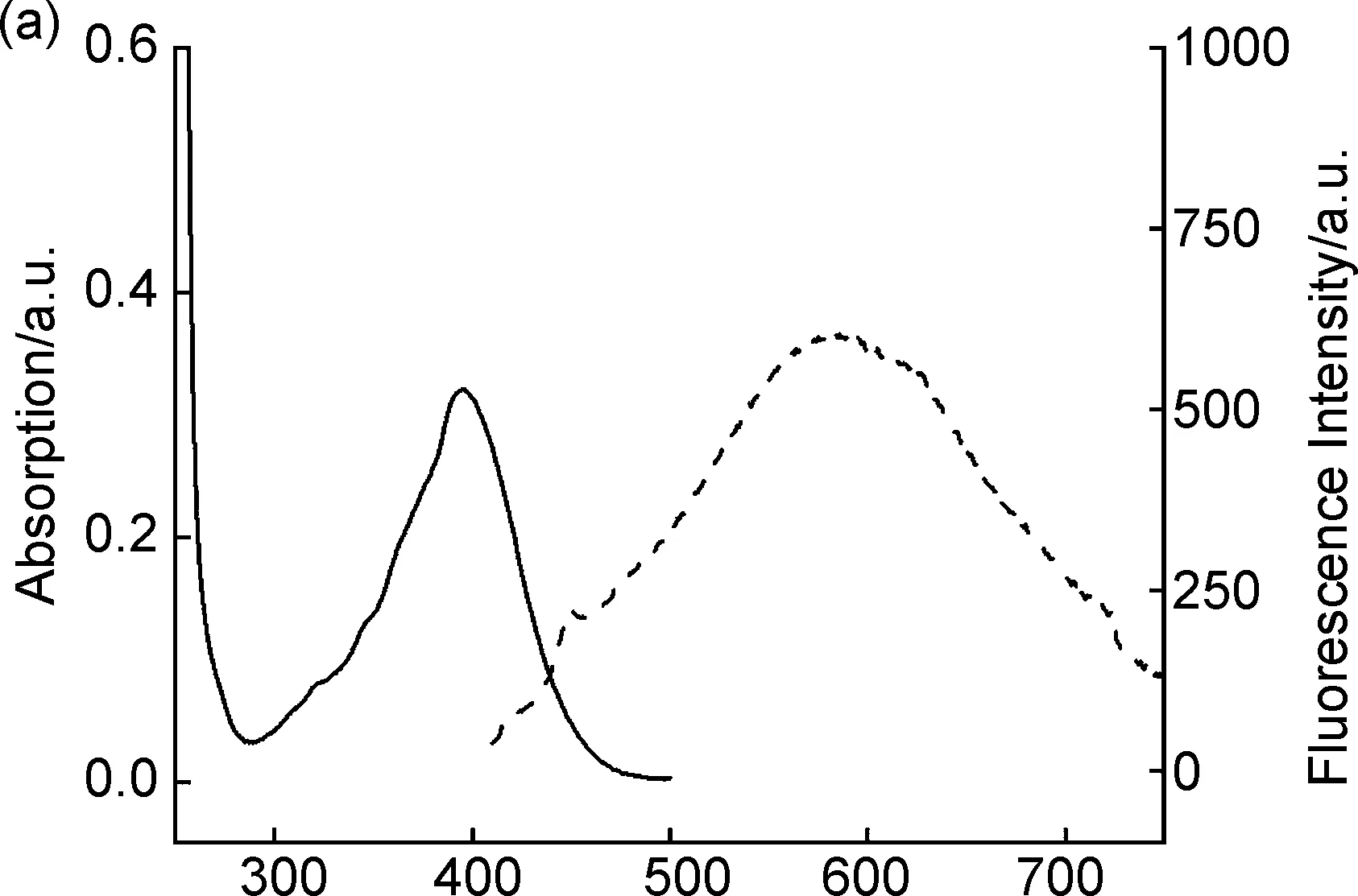
紫外-可見光譜可有效表征化合物在紫外至可見光波段的吸收性能,熒光光譜則表征化合物在紫外或可見光照射下發射電磁波的性能。紫外光譜和熒光光譜在發光材料和染料等領域發揮著重要作用,能對化合物的發色團提供很好的表征信息[15]。由于合成的化合物1a和1b具有較大的共軛體系,因此,其紫外最大吸收波長和熒光的最大發射波長會紅移。化合物1a和1b的紫外(實線)和熒光光譜(虛線)如圖3所示,1a在乙醇中的最大吸收波長為396 nm;以396 nm為激發波長,測得熒光光譜的最大發射波長為583 nm(圖3a)。化合物1b的最大吸收波長為395 nm,熒光光譜的最大發射波長為574 nm(圖3b)。從結構上看,化合物1a和1b具有的最大吸收波長和最大發射波長主要歸因于蒽環、α,β-不飽和酮和氮芥結構p軌道電子的大共軛體系。
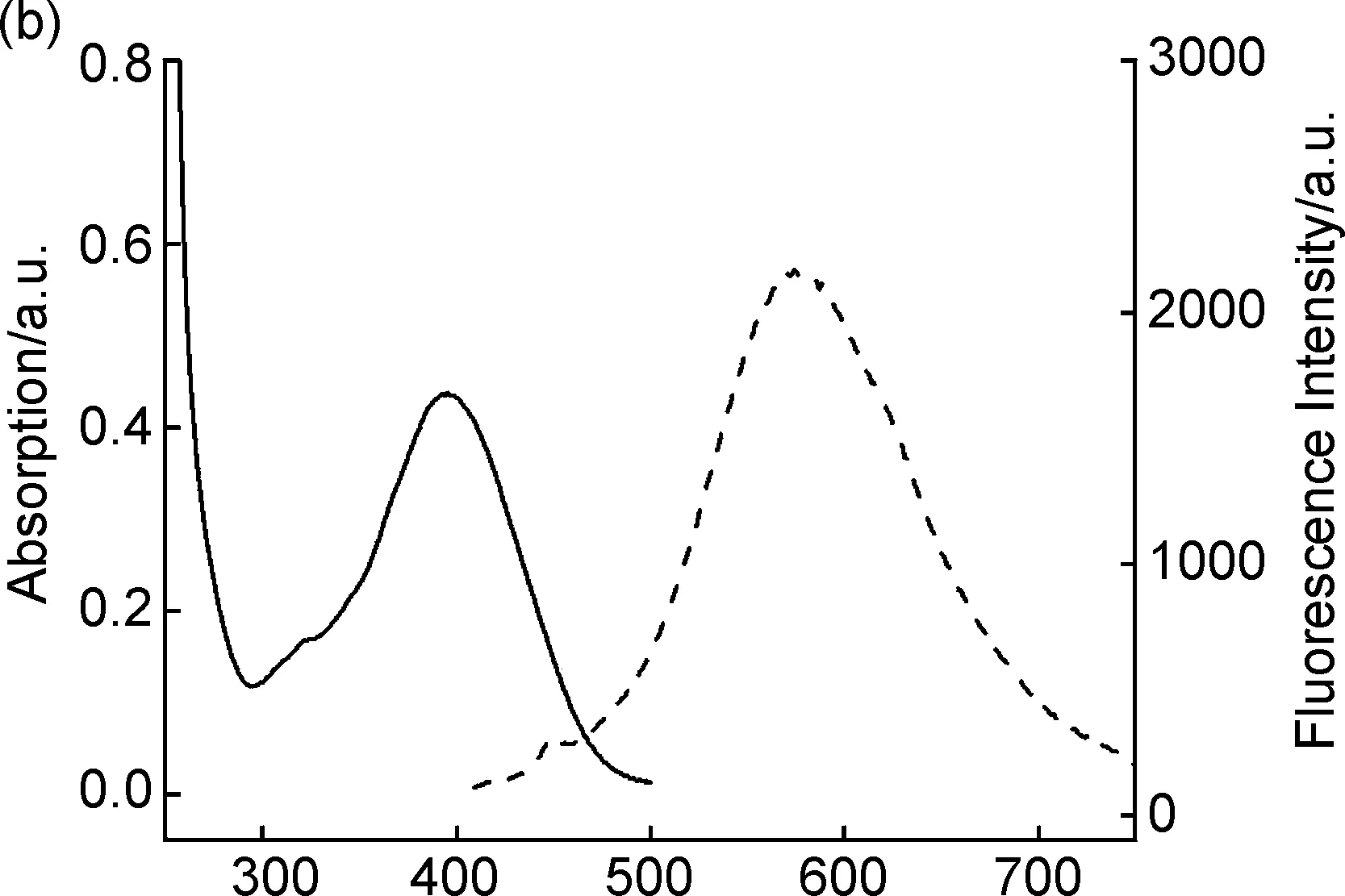
λ/nm圖3 化合物1a(a)和1b(b)在乙醇中的紫外吸收光譜(實線)和熒光光譜(虛線)
2.3 體外抗腫瘤活性
化合物1a和1b對A549、 786-O、 Hela和MDA-MB-231腫瘤細胞增殖的抑制活性見圖4,半抑制濃度(IC50, μM)見表2。由圖4和表2可知,2個化合物對Hela細胞的抑制效果最好。其中1a對上述細胞的半抑制濃度(IC50)為28.8 μM、 23.5 μM、 18.3 μM和27.1 μM。1b對腫瘤細胞的IC50值分別為33.4 μM、 27.1 μM、 24.9 μM和27.5 μM。以上結果證實,2個化合物1a和1b對腫瘤細胞具有良好的抗增殖活性,并且化合物1a的抗增殖活性略優于1b。
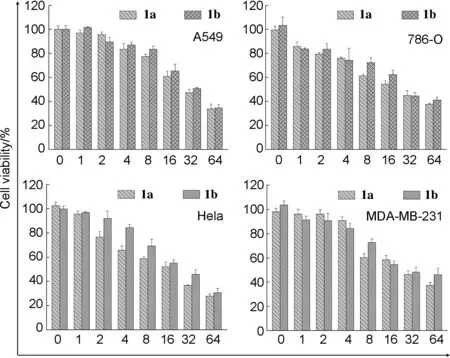
c/μM圖4 化合物1a和1b對腫瘤細胞A549、 786-O、 Hela和MDA-MB-231的抗增殖活性
2.4 化合物1a的抗腫瘤轉移作用
轉移是惡性腫瘤的基本特性之一,很多腫瘤治療失敗的主要原因是因為轉移。因此相對腫瘤的轉移而言,預防比治療更重要。細胞劃痕實驗可以體現不同處理組的細胞遷移的能力。圖5a中僅經過24 h,不加藥組的Hela細胞表現出非常強的遷移能力,而加藥組的細胞遷移則明顯受到抑制,并隨著濃度增加,抑制效果更為顯著,呈現濃度依賴關系。對應統計圖見圖5b,經過24 h后,不加藥組的細胞相對遷移距離為49.5%。當化合物1a加藥濃度分別為20 μM時,相對遷移距離百分數為74.6%,可知化合物1a明顯抑制了Hela細胞的遷移。當濃度為40 μM時,相對遷移距離百分數為83.2%,Hela細胞的遷移進一步被1a抑制。由以上結果可知,化合物1a可通過降低腫瘤細胞的轉移能力來發揮抗腫瘤作用,具有很好的抗腫瘤轉移作用。

表2 化合物1a和1b對腫瘤細胞的半抑制濃度(IC50, μM)

2.5 化合物1a對Hela細胞周期的影響
細胞周期分布可以體現細胞每個時期的比例,進而反應藥物對周期的影響[16]。由圖6可知,S期的比例隨著加藥濃度的增加而增加。不加藥組的G1、 S和G2/M期的比例分別為54.1%, 30.8%和15.1%;當1a濃度為20 μM時,G1期的比例下降為20.9%。而S和G2/M期則分別升高到49.9%和29.2%;當濃度上調到40 μM時,G1期的比例進一步下降到12.3%, S期的比例顯著升高到67.7%,而G2/M期的比例降低到20.0%。由此可知,S期的比例升高總體上呈濃度依賴關系,同時G1和G2/M期沒有明顯的增加,說明化合物1a能有效使Hela細胞阻滯于S期而發揮抗增殖作用。
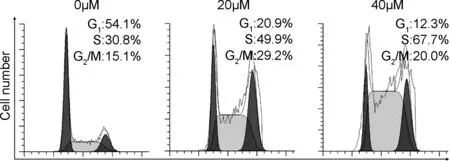
DNA Content圖6 化合物1a對Hela細胞周期的影響
本文運用Vilsmeier-Haack反應和羥醛縮合反應,合成了2種含氮芥和蒽環的新型α,β-不飽和酮,兩步反應都具有較高的產率。從體外抗腫瘤實驗結果可知:1a和1b具有良好的抗腫瘤活性,能有效地抑制多種腫瘤細胞的增殖。其中,1a較1b的抗增殖活性稍強。1a具有優異的抗腫瘤轉移作用,并且通過使Hela細胞的周期阻滯在S期而發揮抗增殖作用。
