
釕(Ru)修飾的鋯基金屬有機框架材料(Zr-MOF)催化酰胺化反應
2023-08-10 03:29:52黃飛鴻王樂張朋飛
山東化工 2023年11期
關鍵詞:催化劑
黃飛鴻,王樂,張朋飛
(東華大學 材料科學與工程學院 纖維材料改性國家重點實驗室,上海 201600)
含氮分子是大部分生物活性分子、藥物合成中間體、天然產物和功能性材料的重要組成單元[1-3]。酰胺化反應作為最古老的有機化學反應之一,能夠準確地在分子內引入氮原子,酰胺基團具有獨特的氫鍵能力,其中普遍存在的酮互變異構體,酰胺基團中就存在兩種不同類型的氫鍵位點,一部分是羰基,其可以作為氫鍵的受體,另一部分則是可以作為氫鍵供體的氨基。從而實現分子的生物活性和藥化性質的導向轉變[4-5]。據統計,市場上在售的醫藥藥物分子排行榜中前200位中,有118種藥物分子含有氮原子。早在2007年,全球制藥公司就把如何實現酰胺鍵的構建列入到搶先研究的項目當中,至此以來,酰胺鍵的構建一直都是有機化學領域亟需解決的研究問題。然而傳統酰胺化反應大多需要較高的反應溫度來促進偶聯反應的進行,但存在著酰氯儲存易分解、化學廢物多、反應條件嚴苛等限制因素[6-7]。而且會浪費大量的縮合試劑,成本過高。隨著研究的不斷深入,通過催化方法得到酰胺類產物,能夠兼顧經濟環保的同時,還能實現酰胺化產物的高效產出。于是愈來愈多的科研人員將目光轉移到合成穩定高效的催化劑降低反應勢壘來催化酰胺化反應進行[8-9]。目前酰胺化反應的催化劑主要是通過貴金屬配合物或者氮雜環卡賓過渡金屬配合物等金屬催化劑催化醛類、醇類和酮類小分子酰胺化。這類方法由于底物范圍較窄、存在金屬離子殘留、產物不易分離以及成本過高等因素大大限制了其在工業領域的應用。如何克服以上不足,構建一個穩定高效的催化反應體系實現酰胺的高效合成是一個亟需解決的研究問題。
金屬有機框架材料(MOFs)是一類由金屬離子/團簇和高度對稱的有機配體,通過金屬與配位原子的強配位作用生成的一類具有永久空隙的晶態多孔材料[10-12]。MOFs不僅有精確的微化學環境,明確的活性位點,還具有豐富的結構預設計性,可以通過對金屬離子/團簇和配體的調控以及對MOFs的合成后修飾來得到一系列具有特定功能的MOFs。如今,人類日常生活中用到的各種化學產品95%以上(按體積計)是通過工業生產和加工出來的,而催化劑是工業生產和產品加工過程中最為關鍵的因素,這使得對更高效催化材料的研究成為一個激動人心且充滿活力的研究領域。金屬有機框架材料由于其前所未有的結構多樣性、固有的有機雜化-無機性質、存在不協調的金屬位置和易于接近的有機支柱、合理設計的可能性以及明確的孔隙度等特點,使成為了非均相催化劑設計的最佳平臺。一般來說金屬有機框架材料可以通過以下幾個位置進行催化作用:1)當金屬氧簇出現空配位點時,可以通過材料裸露的金屬位點進行催化;2)通過配體的設計引入催化位點;3)利用雜化材料光電屬性和配體與金屬間電荷轉移來觸發光催化過程;4)作為其他催化位點(如納米顆粒、酶或其他部分)封裝的宿主;5)通過MOF支架的合成后改性;6)作為通過控制分解形成納米顆粒或單中心催化劑的前體;以及通過結合上述幾個方式進行催化劑的設計。因此,MOFs逐漸成為了非均相催化劑設計與構建的最佳平臺,而鋯基金屬有機框架材料(Zr-MOFs)由于Zr-O鍵的強配位作用,使得Zr-MOFs表現出更加優異的熱穩定性和化學穩定性[13]。光催化由于其清潔、無污染等優點,且光敏劑小分子能夠在光激發下躍遷至激發態通過電子轉移等方式實現小分子高效氧化。基于以上等優點,本文通過對鋯基金屬有機框架材料(Zr-MOFs)進行后修飾改性并作為非均相光催化劑,光催化氧化醛類分子原位生成酰氯活性中間體,通過簡單的一鍋法反應,高效合成酰胺衍生物。
1 實驗部分
1.1 催化劑合成
光催化劑小分子[Ru(dcbpy)(bpy)2]Cl2的合成首先在裝有冷凝器和磁力攪拌棒的100 mL的雙頸圓底燒瓶中加入三氯化釕(RuCl3,400 mg,1.92 mmol)、2,2′-聯吡啶(bpy,600 mg,3.85 mmol)和氯化鋰(LiCl,800 mg,18.87 mmol)。循環抽真空通氮氣三次后,加入13 mL DMF溶液。將反應混合物在N2下回流24 h,反應完成后加入丙酮(120 mL)。最后,將混合物放在冰箱中過夜,得到粗黑綠色晶體(cis-Ru(bpy)2Cl2·2H2O)。過濾混合物并用去離子水和乙醚洗滌。然后,160 mg,0.33 mmol產物和 2,2′-聯吡啶-5,5′-二羧酸(H2dcbpy,101 mg,0.42 mmol)混合在20 mL EtOH/H2O(體積比)中,在N2條件下回流24 h,通過真空濃縮除去溶劑后,在甲醇/乙醚混合溶液中重結晶得到[Ru(dcbpy)(bpy)2]Cl2,如圖1所示。1H NMR(600 MHz,DMSO-d6):δ9.05(d,J= 8.5 Hz,1H),8.91(dd,J= 24.5,8.1 Hz,2H),8.53(dd,J= 8.4,1.9 Hz,1H),8.24(td,J= 7.9,1.5 Hz,1H),8.18(td,J= 7.9,1.5 Hz,1H),8.01(d,J= 1.8 Hz,1H),7.88~7.84(m,1H),7.81~7.77(m,1H),7.61(dd,J= 7.5,5.8 Hz,1H),7.51(dd,J= 7.5,5.8 Hz,1H)。
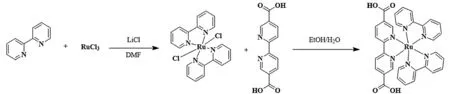
圖1 [Ru(dcbpy)(bpy)2]Cl2的合成
MOFs的合成方法是根據已報道過的合成方法進行了適當的修改[14],PCN-808合成先稱取四氯化鋯(ZrCl4,60 mg,0.25 mmol)和配體(H4TPTB,30 mg,0.04 mmol),置于15 mL的耐高溫玻璃瓶中。再加入酸性調節劑苯甲酸(2 g,16.39 mmol)和10 mLN,N-二甲基甲酰胺(DMF),放入90 Hz的超聲波清洗機均勻分散20 min,待溶液完全澄清后移入到120 ℃高溫烘箱中,3 d后,可以在常規光學顯微鏡下觀察到針狀無色透明晶體。待冷卻至室溫后,每天用10 mL DMF沖洗3次洗3 d,再用10 mL丙酮(Aceton)每天洗3次洗3 d,在80 ℃下抽真空10 h活化樣品備用。
采用溶劑輔助的后修飾手法對合成后的MOFs材料進行光化學改性。取100 mg PCN-808于玻璃瓶中,另取40 mg的光催化劑小分子[Ru(dcbpy)(bpy)2]Cl2浸泡在20 mL的DMF溶液中 ,分別在裝有MOFs的玻璃瓶中加入10 mL混合溶液,置于100 ℃的高溫烘箱中,通過溶劑的動態吸附光催化劑小分子會與框架中未配位的Zr-O簇配位,從而固定在晶體框架內部,24 h 后取出。用新鮮的DMF溶液清洗3次后得到相應的PCN-808-Ru晶體有機框架材料,并對其進行下一步分析于表征。
1.2 催化劑表征
對合成的PCN-808以及后修飾的PCN-808-Ru進行了粉末X射線衍射分析(PXRD),從而確定樣品的晶體構型,如圖2a所示。通過實驗得到的PXRD衍射圖譜可以看出,實驗合成的晶體以及通過溶劑輔助插入光催化劑小分子完成后修飾的晶體材料,與之前報道的晶體原型具有相同的結晶度,僅僅是相位發生了小小的偏移,由此可以證明,由后修飾法引入光催化位點并不會對晶體的結晶度產生過大的影響。為了進一步驗證經過后修飾過程之后,光催化劑小分子[Ru(dcbpy)(bpy)2]Cl2是否成功地引入到晶體框架內部中,對后修飾后的材料進行了紫外近可見分析,首先根據相關報道,PCN-808晶體材料是沒有光吸收的,如果成功地將配體分子引入的話,那么后修飾得到的PCN-808-Ru應該在[Ru(dcbpy)(bpy)2]Cl2對應的波長范圍出現吸收特征峰,于是優先對光催化劑小分子[Ru(dcbpy)(bpy)2]Cl2,和MOFs材料進行了光譜分析(UV-vis),如圖2b所示。由圖可以明顯看出,在經過后修飾處理得到的PCN-808-Ru相比與未處理的PCN-808在~475 nm左右出現了明顯的吸收峰,而吸收峰的藍移可能是因為晶體的空間結構和化學環境導致光敏劑分子構型發生改變。吸收峰的出現說明通過簡單的溶劑法是可以很好地構建小分子[Ru(dcbpy)(bpy)2]Cl2與晶體框架上位配位的氧原子相互配位,從而通過配體的插入實現光催化位點的成功引入的。

(a)PXRD圖譜;(b)Uiv-vs圖譜圖2 PCN-808和PCN-808-Ru的PXRD圖譜和Uiv-vs圖譜
1.3 條件篩選
首先選取苯甲醛作為反應條件優化的模型底物,從反應時間,催化劑負載量,催化劑種類,氯源含量和光照等因素對最佳反應條件進行篩選,如表1所示。首先對反應時間進行了研究,分別在反應3,5,7 h分別取上清液做產率分析,結果表明在反應7 h效果最佳,如條目1~3所示。之后對催化劑用量和種類進行了對比分析,選取合成的小分子均相催化劑Ru(dcbpy)(bpy)2和PCN-808-Ru進行對比實驗,實驗結果表明當PCN-808-Ru的用量減少到5 mg(物質的量分數約3%時)的酰氯產率高達95%,而相同比例的傳統均相小分子催化劑僅為88%,如條目3~5、8、9所示。其次對氯代試劑NCS的用量進行了研究,根據實驗結果表明NCS的用量在1.5和3 mmol時,產率相當,但出于成本和環保角度考慮NCS用量在1.5 mmol最佳,如條目3、6~7所示。之后對有無光照,有無催化劑進行對比分析,發現幾乎無產物生成,如條目10,11所示。綜上所述,PCN-808-Ru催化酰胺化反應的最佳條件是:底物0.5 mmol,PCN-808-Ru(物質的量分數3%),NCS(1.5 mmol),MeCN(2 mL)反應7 h后旋干反應液,加入芐氧胺鹽酸鹽(3 mmol)和K2CO3(3 mmol)在4 mL DCM中反應16 h得到最終產物芐氧酰胺類衍生物。

表1 PCN-808-Ru催化酰胺化反應條件優化圖
2 結果與討論
通過反應條件篩選得到最優反應條件之后,進一步對底物適用范圍進行了拓展,包括雜原子鄰位、富電子基團、缺電子基團取代的芳香醛類底物進行了反應研究,都取得較高的產率,底物范圍拓展如圖3所示。該反應體系對具有富電子基團取代的芳香醛類底物具有較高的產率,其中苯甲醛(1)和對甲基苯甲醛(2)的產率都高于80%,而具有缺電子基團取代的芳香醛的產率稍低于富電子基團。而對于缺電子基團的強弱對該反應體系的產率沒有過大的影響。對氰基苯甲醛(4)、4-F-苯甲醛(5)、3-三氟甲基苯甲醛(6)和4-氯苯甲醛(7)的產率分別為73%,75%,62%和68%。綜上所述,實驗結果表明該反應體系對芐氧胺類鹽酸鹽和芳香醛的酰胺化反應具有高反應活性和官能團耐受性。

圖3 底物范圍拓展圖
N-(benzyloxy) benzamide(1):由快速柱色譜純化系統分離得到白色固體(洗脫劑:V正己烷∶V乙酸乙酯=8∶2)。1H NMR:δ11.81(s,1H),7.79~7.75(m,2H),7.57~7.52(m,1H),7.50~7.44(m,4H),7.43~7.38(m,2H),7.38~7.34(m,1H),4.95(s,2H)。13C NMR:δ164.44,135.94,132.37,131.63,128.95,128.48,128.35,128.33,127.13,77.03,40.06。
N-(benzyloxy)-4-methylbenzamide(2):由快速柱色譜純化系統分離得到白色固體(洗脫劑:V正己烷∶V乙酸乙酯=8∶2)。1H NMR:δ8.84(s,1H),7.57(d,J= 8.0 Hz,2H),7.45~7.40(m,2H),7.40~7.32(m,3H),7.18(d,J= 7.9 Hz,2H),5.00(s,2H),2.37(s,3H)。13C NMR:δ142.71,135.46,129.45,129.43,129.13,128.86,128.73,127.18,78.43,21.61。
N-(benzyloxy)-3-methylbenzamide(3):由快速柱色譜純化系統分離得到白色固體(洗脫劑:V正己烷∶V乙酸乙酯=8∶2)。1H NMR:δ8.12(s,1H),7.43(d,J= 7.2 Hz,3H),7.39~7.36(m,3H),7.35~7.30(m,1H),7.29~7.23(m,1H),7.18(d,J= 7.5 Hz,1H),5.22(s,2H),2.37(s,3H)。13C NMR:δ149.40,138.53,137.64,132.22,130.84,128.70,128.58,128.51,128.09,127.59,124.61,76.49,21.44。
N-(benzyloxy)-4-cyanobenzamide(4):由快速柱色譜純化系統分離得到白色固體(洗脫劑:V正己烷∶V乙酸乙酯=8∶2)。1H NMR:δ12.04(s,1H),8.10~7.79(m,4H),7.49~7.29(m,5H),4.95(s,2H)。13C NMR:δ162.86,136.37,135.73,132.58,129.01,128.42,128.38,127.98,118.26,114.00,77.10。
N-(benzyloxy)-4-fluorobenzamide(5):由快速柱色譜純化系統分離得到白色固體(洗脫劑:V正己烷∶V乙酸乙酯=8∶2)。1H NMR:δ8.87(s,1H),7.71~7.66(m,2H),7.44~7.40(m,2H),7.40~7.34(m,3H),7.06(t,J= 8.6 Hz,2H),5.00(s,2H)。13C NMR:δ165.97,164.29,135.29,129.63,129.46,128.99,128.80,128.18,115.93(d,J= 22.0 Hz),78.54。
N-(benzyloxy)-3-(trifluoromethyl) benzamide(6):由快速柱色譜純化系統分離得到白色固體(洗脫劑:V正己烷∶V乙酸乙酯=8∶2)。1H NMR:δ8.16(s,1H),7.85(s,1H),7.75(d,J= 7.8 Hz,1H),7.61(d,J= 7.8 Hz,1H),7.49(t,J= 7.8 Hz,1H),7.44~7.41(m,2H),7.41~7.37(m,2H),7.36~7.32(m,1H),5.24(s,2H)。13C NMR:δ147.69,137.32,133.24,131.47,131.25,130.33,129.32,128.65,128.58,128.26,126.41(d,J= 3.5 Hz),123.81(d,J= 3.9 Hz),76.89。
N-(benzyloxy)-2,4-dichlorobenzamide(7):由快速柱色譜純化系統分離得到白色固體(洗脫劑:V正己烷∶V乙酸乙酯=8∶2)。1H NMR:δ8.51(s,1H),7.85(d,J= 8.5 Hz,1H),7.47~7.38(m,6H),7.28~7.21(m,1H),5.25(s,2H)。13C NMR:δ145.20,137.23,136.15,134.43,129.72,128.72,128.63,128.56,128.25,128.04,127.54,76.93。
3 結論
采用溶劑輔助的后合成修飾法,對PCN-808進行光化學改性,并將PCN-808-Ru作為非均相光催化劑催化苯甲醛和芐氧胺鹽酸鹽的酰胺化反應。實驗結果顯示,該反應體系具有較高的產率以及對富電子和缺電子基團的芳香醛都具有較好的耐受性。該項研究結果拓寬了酰胺化產物的合成方法,后合成改性可以很好地對MOFs材料進行導向改性,從而得到具有特定功能的MOFs材料,對推動MOFs材料在催化化學方面的應用具有重大意義。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
