
Fe-MOF催化芳烴C(sp2)-H胺化反應研究
2023-08-10 03:29:54洪文杰王樂
山東化工 2023年11期
關鍵詞:催化劑
洪文杰,王樂
(東華大學 材料科學與工程學院 纖維材料改性國家重點實驗室,上海 201600)
含氮化合物已成為藥物化學、制藥、天然產物合成、有機材料和催化劑領域的關鍵組成部分[1]。其中,芳基胺是許多天然產物和合成化合物中的關鍵成分,具有重要的化學、生物和藥用特性[2]。分子內脫氫釋放H2實現C-H胺化是一種非常有效的結合C-H鍵和N-H鍵的方法。理想情況下,芳香族的C-N鍵可以直接從相應的芳烴和胺中構建,只產生H2作為副產物[3]。但是這類反應存在幾個問題使得轉變并不是直接的[4]。首先,動力學和熱力學屏障的存在使得反應無法自發進行,芳烴中C-H鍵的低極性以及高的pKa值進一步促成了它們的低反應性,且在底物中存在的強度和酸度相似的C-H鍵,就會發生區域選擇性的問題。其次,兩個耦合化合物都具有親核性,導致極性不匹配,進一步阻礙了反應的進行。因此長期以來,人們一直在努力開發高效的反應,將含氮基團引入芳烴分子。
自從過渡金屬催化的交叉偶聯反應發展后,現代有機合成取得了相當大的進展[5]。傳統上將氨基引入芳烴中最重要的方法是親電芳香族硝化,然后還原硝基[6]。然而,芳烴的硝化通常需要強酸性或氧化反應條件,這限制了其在具有敏感官能團的底物上的使用。過渡金屬催化的C-N交叉偶聯反應是一種通過C-N鍵形成將氮引入有機分子的通用方法,它擴大了合成范圍,縮短了合成程序。利用過渡金屬催化C-N鍵的形成的方法之一是Buchwald-Hartwig胺化反應類交叉耦合反應[7-8]。該反應在鈀和堿的存在下,胺化合物與芳基鹵素化合物發生交叉偶聯反應,形成C-N鍵,得到胺的N-芳基化合物。但這種交叉耦合反應需要在胺化反應前引入鹵化物基團,且在反應后生成鹵化物鹽等副產物[9]。采用C-H鍵的胺化是催化C-N鍵形成的另一種方法,無需預安裝反應基團,直接使碳氫化合物的底物官能化[10]。但由于C-H鍵的鍵能較高,pKa較低,因此C-H鍵的活化仍有一定的難度。將無處不在的C-H鍵催化轉變為具有高經濟價值的C-N鍵,是合成含N分子有效的合成方案。胺類化合物能通過C-H鍵的活化從芳烴合成,是合成芳基胺化合物的最具步驟和原子經濟性的方法之一。
分子間直接胺化反應是一種強大而有力的合成工具,因為不需要合成制備所需要的底物。形成C-N鍵的交叉脫氫偶聯仍然是最理想的,因為它避免了預功能化步驟。考慮到催化劑的經濟和環境效益,在C-H功能化過程中使用廉價過渡金屬催化劑是一種有吸引力的策略。金屬催化的C-H鍵直接胺化反應中已經使用了多種胺化試劑[11]。盡管非官能團前化的胺或酰胺是最理想的氮源,但需要外部氧化劑才能與這些反應物進行催化C-H胺化反應。在金屬催化劑的作用下,從胺化試劑衍生而出的帶有極化N-X鍵的化合物能夠發生氧化裂解[12]。因此這類胺化試劑可以在不額外添加氧化劑的情況下,也作為內部的氧化劑使用。雖然在實現芳族C-H胺化方面有了重大突破,但在適用的芳族化合物范圍內仍然存在嚴重的局限性。由于獲得反應性普遍困難,許多報道的反應僅適用于具有催化劑配位基團或電子活化的芳香族化合物。因此,開發用于簡單芳烴的C-H胺化反應和催化劑一直是該領域的一項艱巨任務。
在過去的研究中,晶態多孔MOF材料因其結構可設計性、功能可設計性以及較好的物理以及化學穩定性等,已經被廣泛地研究并應用于非均相催化領域[13]。在催化方面的研究MOF的晶體結構決定了其主要側重于非均相催化。由于MOF的組成和結構特征的結合,MOF被認為是非均相催化中最有前途和最通用的材料之一。MOF的催化活性可以歸因于其各種物理化學性質,例如高比表面積、大孔徑、足夠的框架穩定性、尺寸和形狀的可調節性、易于回收、浸出少等[14]。MOF的這些特性使得在高活性下又具有高選擇性,具有均相以及非均相催化劑的整體優勢,使其在非均相催化中更有吸引力。與均相催化劑相比,MOF非均相催化劑可以在催化反應結束后回收使用,這解決了均相催化劑難以回收使用等難題。與傳統的微孔以及介孔材料相比,高度的孔隙率和廣闊的比表面積以及有機和無機成分的變化性為MOF的結構和性質提供了很大程度的多樣性。得益于MOF擁有的精確晶態結構,因此它在非均相催化方面對催化活性位點的研究變得明確可行。MOF在C-H鍵的催化活化方面具有幾個特點,如催化活性位點分布均勻、能牢固固定活性位點、金屬和配體具有可修飾性以及可用單晶衍射儀精確確定空間結構表征催化活性位置[15]。這些都有利于MOF催化C-H鍵活化機理的研究。在數不清的MOF材料中,鐵基金屬有機框架具有結構多樣性、毒性低、穩定性好、功能設計等諸多特性,在實際應用中具有極佳的潛力,受到了廣泛的研究[16-17]。因此我們希望探索使用廉價鐵基金屬有機框架材料作為非均相催化劑,催化C-H鍵的活化反應。在此我們報道了使用廉價金屬鐵基金屬有機框架材料Fe-MOF作為非均相催化劑,使用親電胺化試劑作為胺源,以羥胺衍生物催化芳烴C-N鍵的形成反應。胺化試劑的N-O鍵相對較弱,在催化胺化過程中裂解時作為內部氧化劑的作用,從而避免了向反應混合物中添加外部氧化劑的需要。反應條件溫和,無需對底物預安裝導向官能團,成功實現簡單烷烴取代的芳烴化合物C(sp2)-H胺化。
1 實驗部分
1.1 Fe-MOF催化劑合成及表征
Fe-MOF的合成參考了先前文獻報道的方法[18]。首先將四水合氯化鐵(0.04 g,0.3 mmol)、1,3,5-苯三甲酸(0.1 g,0.45 mmol)溶解在N,N-二甲基甲酰胺(DMF,4.5 mL)和冰乙酸(1.5 mL)的混合溶液中。分散溶解均勻后,將混合反應溶液轉移至水熱釜中。采用溶劑熱方法,于150 ℃烘箱中反應36 h。反應結束后,可以得到黃色塊狀晶體。將獲得的晶體用DMF清洗3 d,每天清洗三次以除去未反應的原料。 用甲醇清洗3 d,每天更換三次新鮮甲醇溶液。將活化后的Fe-MOF在真空下干燥,獲得深黃色塊狀晶體。粉末X射線衍射法是表征晶體結構強有力的手段之一,常用于晶體結構的物相分析。將使用水熱法合成的Fe-MOF進行XRD分析測試,結果如圖1所示。合成的Fe-MOF的衍射峰與文獻中報道的圖譜吻合。表明成功合成了晶態多孔材料Fe-MOF。

圖1 Fe-MOF的XRD譜圖
1.2 胺化試劑合成及表征
胺化試劑NsONH2MeOTf合成步驟如圖2所示。
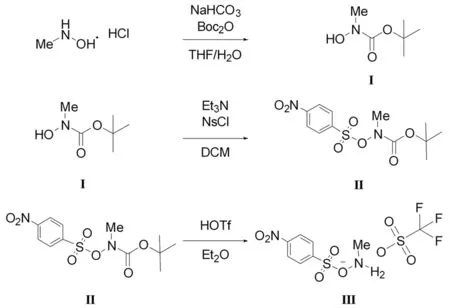
圖2 胺化試劑合成反應式
將N-甲基羥胺鹽酸鹽(5.0 g,59.9 mmol,1.0 mol/L)溶解于四氫呋喃(100 mL)和水(10 mL)的混合溶液中,加入碳酸氫鈉(10.0 g,119.0 mmol,2.0 mol/L)和二碳酸二叔丁酯(15.7 g,72.0 mmol,1.2 mol/L),室溫下攪拌16 h。反應結束后,將反應液用水(50 mL)稀釋,并用二氯甲烷(3×50 mL)萃取。合并的有機相用無水硫酸鎂干燥,真空濃縮得橙色油狀混合物。對獲得的橙色油狀混合物進行柱色譜分離純化,在V(正己烷)∶V(乙酸乙酯)=3∶7洗脫劑下獲得無色油狀化合物Ⅰ(NMeOHBoc,6.7 g,45.6 mmol,76%)。1H NMR(600 MHz,Chloroform-d)δ3.17(s,3H),1.50(s,9H)。13C NMR(151 MHz,Chloroform-d)δ157.61,81.86,37.78,28.29。
將化合物Ⅰ(6.7 g,45.6 mmol,1.0 mol/L)溶解在二氯甲烷中(90 mL)。將溶液冷卻至0 ℃后,加入三乙胺(4.6 g,45.6 mmol,1.0 mol/L),隨后加入對硝基苯磺酰氯(10.17 g,46.0 mmol,1.0 mol/L)。將反應液升至室溫并攪拌16 h。反應結束后,有機相用去離子水(3×50 mL)、碳酸氫鈉溶液(3×50 mL)和飽和食鹽水(3×50 mL)洗。合并的有機相用無水硫酸鎂干燥,減壓干燥獲得灰白色固體Ⅱ(NsONMeBoc,12.6 g,38.0 mmol,83%)。無需進一步純化即可進行下一步使用。1H NMR(600 MHz,Chloroform-d)δ8.44~8.39(m,2H),8.24~8.19(m,2H),3.31(s,3H),1.24(s,9H)。13C NMR(151 MHz,Chloroform-d)δ155.59,151.14,139.80,131.12,123.90,84.19,40.75,27.65。
將灰白色固體Ⅱ(12.6 g,38.0 mmol,1.0 mol/L)懸浮在乙醚(120 mL)中,將所得的懸浮液冷卻至0 ℃,在劇烈攪拌下滴加三氟甲烷磺酸(5.7 g,38.0 mmol,1.0 mol/L)。添加完成后,使反應液升至室溫并反應2 h。反應結束后,過濾固體,用冰乙醚(3×20 mL)洗滌,干燥得白色固體胺化試劑Ⅲ(NsONH2MeOTf,9.0 g,23.6 mmol,62%)。1H NMR(600 MHz,Acetonitrile-d3)δ9.54(s,2H),8.47~8.43(m,2H),8.25~8.20(m,2H),2.90(s,3H)。13C NMR(151 MHz,Acetonitrile-d3)δ152.71,140.48,131.60,125.66,39.69。19F NMR(565 MHz,Acetonitrile-d3)δ-79.50。
1.3 反應條件篩選
選取了均三甲苯作為反應條件優化實驗的底物模型,以合成的試劑NsONH2MeOTf為胺化試劑,合成的Fe-MOF作為非均相催化劑,對反應溶劑、反應溫度、催化劑用量等方面進行了條件篩選,如表1所示。首先篩選了在室溫下反應溶劑對反應的影響,發現該N-甲基苯胺化反應高度依賴于1,1,1,3,3,3-六氟-2-丙醇(HFIP),在其余溶劑中皆無產物出現。推測是所使用的胺化試劑不溶于HFIP外的溶劑,觀察反應現象,只有HFIP溶劑能充分溶解胺化試劑NsONH2MeOTf,其余溶劑不能溶解或者不完全溶解胺化試劑NsONH2MeOTf。初步確認反應溶劑后,調整反應溫度,在40 ℃達到了94%的產率。進一步在40 ℃反應條件下優化催化劑Fe-MOF的使用量,當使用量為5 mg時,產率在93%,使用量15 mg時,產率在89%。繼續提高催化劑的用量并不能較好提高反應產率。因此將催化劑Fe-MOF的使用量定為10 mg。

表1 Fe-MOF催化C(sp2)-H胺化反應條件優化
綜上,Fe-MOF催化芳烴的C(sp2)-H胺化最優反應條件是:Fe-MOF(10 mg),溶劑為HFIP溶劑,40 ℃下反應3 h。
2 結果與討論
在得到最優反應條件后,進一步對底物適用范圍進行了拓展,底物范圍拓展如圖3所示。三烷基取代的均三甲苯(1)以及1,3,5-三乙基苯(2)獲得了較高的產率,產率分別為94%,90%。對于簡單的單烷基取代苯,反應具有中等的收率,對于甲苯(3)和乙苯(4)的產物,都只有單一的對位取代的N-甲基苯胺衍生物,產率分別為75%,56%。而單取代基是大位阻基團的叔丁基苯(5)則出現鄰位和對位取代的兩種N-甲基苯胺衍生物,產率為57%,鄰位和間位產物比為11∶1。對于二烷基取代的間二甲苯(6),反應則表現出較高的產率和較好的選擇性,產率為85%,且只有對應的單取代的產物。實驗表明該體系具有高反應性以及優異的位點選擇性。

圖3 C(sp2)-H胺化底物范圍擴展
N,2,4,6-tetramethylaniline(1):由快速色譜純化系統分離得到黃色液體(洗脫劑:V正己烷∶V乙酸乙酯=9∶1)。1H NMR(600 MHz,Chloroform-d)δ6.89(s,2H),2.90(s,1H),2.80(s,3H),2.33(s,6H),2.29(s,3H)。13C NMR(151 MHz,Chloroform-d)δ144.95,131.37,129.59,129.52,35.66,20.62,18.22。
2,4,6-triethyl-N-methylaniline(2):由快速色譜純化系統分離得到黃色液體(洗脫劑:V正己烷∶V乙酸乙酯=9∶1)。1H NMR(600 MHz,Chloroform-d)δ6.95(s,2H),2.93(s,1H),2.81(s,3H), 2.72(q,J=7.6 Hz,4H),2.64(q,J=7.6 Hz,2H),1.34~1.28(m,9H)。13C NMR(151 MHz,Chloroform-d)δ144.08,138.47,136.40,126.28,37.31,28.45,24.47,15.78,15.16。
N,4-dimethylaniline(3):由快速色譜純化系統分離得到黃色液體(洗脫劑:V正己烷∶V乙酸乙酯=9∶1)。1H NMR(600 MHz,Chloroform-d)δ7.05~7.00(m,2H),6.57(d,J=8.4 Hz,2H),2.83(s,3H),2.27(s,3H)。13C NMR(151 MHz,Chloroform-d)δ147.25,129.80,126.59,112.73,31.21,20.49。
4-ethyl-N-methylaniline(4):由快速色譜純化系統分離得到黃色液體(洗脫劑:V正己烷∶V乙酸乙酯=9∶1)。1H NMR(600 MHz,Chloroform-d)δ7.08(d,J=8.3 Hz,2H),6.62(d,J=8.3 Hz,2H),2.86(s,3H),2.60(q,J=7.6 Hz,2H), 1.24(t,J=7.6 Hz,3H)。13C NMR(151 MHz,Chloroform-d)δ147.39,133.20,128.57,112.65,31.11,28.01,16.10。
4-(tert-butyl)-N-methylaniline(5a):由快速色譜純化系統分離得到黃色液體(洗脫劑:V正己烷∶V乙酸乙酯=9∶1)。1H NMR(600 MHz,Chloroform-d)δ7.28(dd,J=8.8,2.3 Hz,2H), 6.65~6.61(m,2H),2.87(s,3H),1.33(s,9H)。13C NMR(151 MHz,Chloroform-d)δ147.03,140.08,125.99,112.24,33.87,31.59,31.38。
3-(tert-butyl)-N-methylaniline(5b): 由快速色譜純化系統分離得到黃色液體(洗脫劑:V正己烷∶V乙酸乙酯=9∶1)。1H NMR(600 MHz,Chloroform-d)δ7.19(t,J=7.9 Hz,1H),6.82~6.80(m,1H),6.69(t,J=2.1 Hz,1H),6.50(dd,J=8.0,2.4 Hz,1H),2.89(s,3H),1.35(s,9H)。13C NMR(151 MHz,Chloroform-d)δ152.26,149.13,128.91,114.72,110.14,109.34,34.66,31.01,30.91。
N,2,4-trimethylaniline(6):由快速色譜純化系統分離得到黃色液體(洗脫劑:V正己烷∶V乙酸乙酯=9∶1)。1H NMR(600 MHz,Chloroform-d)δ7.02(d,J=8.8 Hz,1H),6.95(s,1H),6.59(d,J=8.1 Hz,1H),3.46(s,1H),2.92(s,3H),2.30(s,3H),2.17(s,3H)。13C NMR(151 MHz,Chloroform-d)δ145.10,130.92,127.47,126.10,122.19,109.45,31.17,20.45,17.46。
3 結論
采用廉價金屬基鐵基MOF材料作為非均相催化劑,催化了簡單芳烴的C(sp2)-H胺化反應,成功對簡單芳烴實現直接胺化生成N-甲基苯胺類衍生物。使用高親電的羥胺衍生物NsONH2MeOTf作為胺化試劑,既提供氮源,又包含容易離去的氧化基團,避免胺化反應中額外添加其他氧化劑。不需要在芳烴底物上預安裝任何導向基團,即可在溫和條件下獲得一系列胺化產物,且反應具有優異的選擇性。催化領域一項合乎演變的邏輯是將可溶性的均相反應轉變為不可溶性的非均相反應。將MOF作為非均相催化劑催化有機反應的進行,并用于構建天然產物中間體有著巨大的應用價值。這為碳氫鍵的活化開辟了新的道路,并且對理解催化反應中的機理研究有著重要作用。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
