普克魯胺中間體3-氟-4-異硫氰酸根-2-三氟甲基苯甲腈的合成
2023-08-19 00:30:04劉雙雙詹樂武李斌棟
合成化學 2023年8期
劉雙雙, 侯 靜, 王 娟, 詹樂武, 李斌棟
(南京理工大學 化學與化工學院,江蘇 南京 210094)
普克魯胺(Proxalutamide,圖1)作為新一代雄激素受體(AR)拮抗劑,對轉移性前列腺癌和轉移性乳腺癌具有良好的治療效果[1-4]。 Proxalutamide是抗前列腺癌新藥恩雜魯胺(Enzalutamide, MDV3100)的核心結構,是采用靶蛋白晶體結構的計算機輔助設計并經反復優化后得到的新型化合物,與恩雜魯胺(MDV3100)相比,其化學結構存在多處改變,從而導致性質產生了較大變化,例如Proxalutamide改善了分子溶解度和動力學性質[5-6]。自2020年新冠疫情爆發后,科學家將Proxalutamide的治療領域應用到新冠病毒方面,研究發現,Proxalutamide對治療新冠輕癥和中癥患者效果顯著,目前仍處于臨床階段[7]。
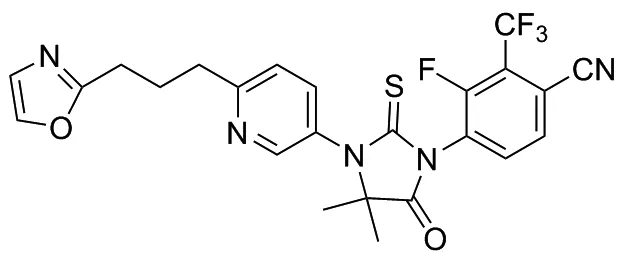
圖1 普克魯胺的結構式
3-氟-4-異硫氰酸根-2-三氟甲基苯甲腈(16)為合成Proxalutamide的重要中間體,而2-氟-3-三氟甲基苯胺(11)為合成Proxalutamide的關鍵前體化合物。目前,國內外關于11的合成工藝的研究報道較少,阻礙了Proxalutamide的大規模生產與應用。1989年,Wollweber[8]報道了以2-氟-5-氯-三氟甲苯為原料,經硝化得到2-氟-5-氯-3-硝基-三氟甲苯,再經催化氫化合成11。然而該工藝條件下的收率僅為50%,并且存在原料不易得、操作條件苛刻和危險系數較高等不利因素,因而不利于工業化生產。2021年,劉輝等[9]以鄰氟三氟甲苯為原料,經疊氮化反應得到2-氟-3-三氟甲基苯基疊氮,再經雷尼鎳、氫氣催化加氫還原合成11。該工藝雖然合成步驟較少,但存在高溫高壓操作條件帶來的潛在危險。
本文設計了一條合成16的新路線(圖2)。以2-氟三氟甲苯為原料,經羧基化反應生成2-氟-3-三氟甲基苯甲酸(1a);1a經酰胺化反應生成2-氟-3-三氟甲基苯甲酰胺(1b);1b經Hofmann重排反應合成2-氟-3-三氟甲基苯胺(11)。11再經溴代合成12,12經氰基取代合成13,14,接著經水解合成15及氨基氧化合成最終產物Proxalutamide中間體3-氟-4-異硫氰酸根-2-三氟甲基苯甲腈(16),總收率36%。該合成路線的所有反應均為常壓反應,操作簡易安全,原料及試劑易于獲取,同時各步反應收率和重復性較好。
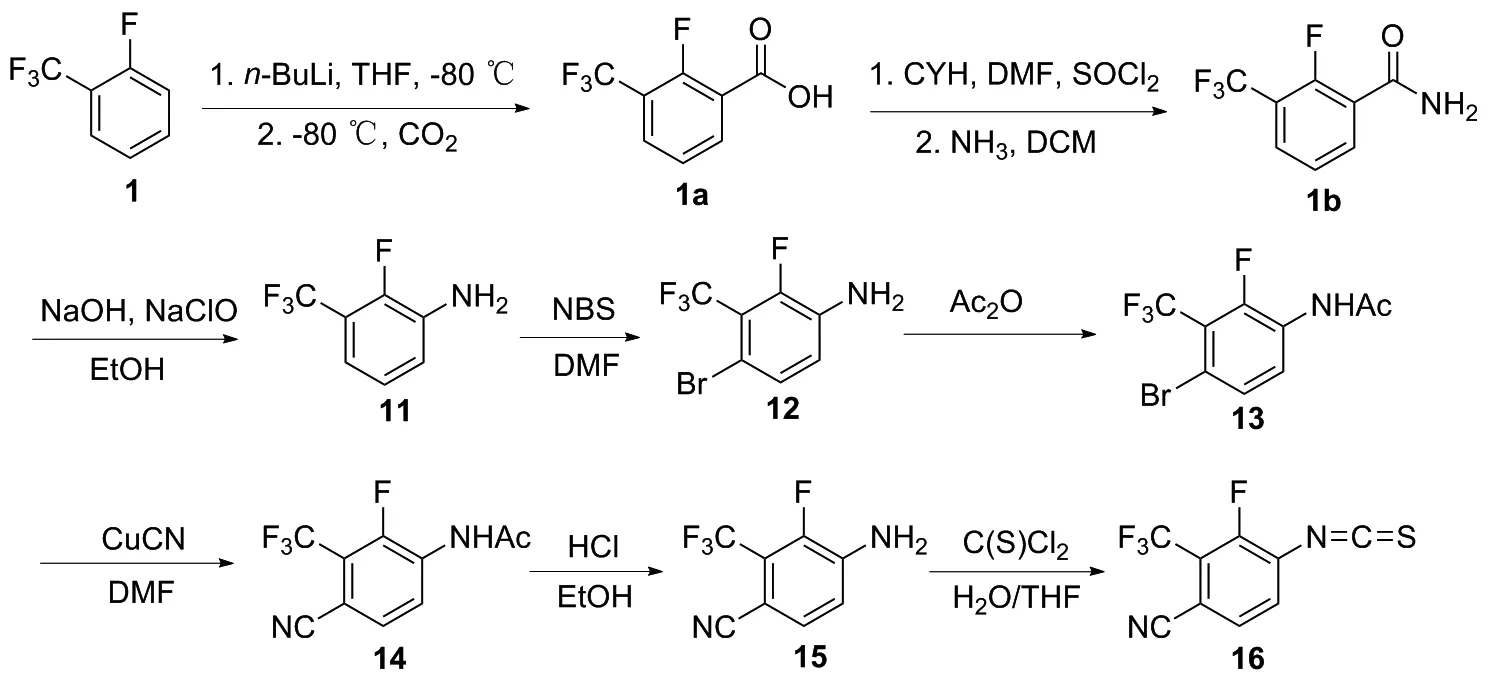
圖2 化合物16的合成路線
1 實驗部分
1.1 儀器與試劑
WRS-1B型數字熔點儀;Bruker Avance 500 MHz型核磁共振儀(TMS為內標,DMSO-d6或CDCl3為溶劑)。
所用試劑均為分析純。
1.2 合成
(1) 2-氟-3-三氟甲基苯甲酸(1a)的合成
向250 mL圓底燒瓶中依次加入鄰氟三氟甲苯(5.00 g, 30.49 mmol)和四氫呋喃(35 mL),氮氣保護。溫度降至-80 ℃,滴加濃度為2.70 mol/L正丁基鋰的正己烷溶液(12.40 mL, 33.52 mmol),滴加完畢后保溫反應5 h,并于-70 ℃條件下向反應體系中通入二氧化碳,鼓泡反應20 min。溫度升高至-30 ℃,加入20 mL去離子水淬滅殘余正丁基鋰,溫度升至室溫,靜置分層,分出水相,水相減壓脫溶至無共沸,加入鹽酸調節pH=2左右,有白色固體析出。于2~8 ℃條件下析晶0.5 h,抽濾,將濾餅于60 ℃條件下干燥,得白色固體化合物1a6.07 g,收率95.6%, m.p.126~128 ℃;1H NMR(500 MHz, DMSO-d6)δ: 13.74(s, 1H), 8.16(t,J=7.5 Hz, 1H), 7.99(t,J=7.0 Hz, 1H), 7.50(t,J=7.8 Hz, 1H);13C NMR(126 MHz, DMSO-d6)δ: 163.93, 159.22, 136.58, 131.29, 124.97, 123.55, 121.38, 121.17。
安徽省秸稈總量約4200萬噸,其中水稻秸稈約1490萬噸,小麥秸稈約1200萬噸、玉米秸稈約400萬噸,大豆秸稈約230萬噸[12]。
(2) 2-氟-3-三氟甲基苯甲酰胺(1b)的合成
向100 mL燒瓶中加入1a(2.00 g, 9.61mmol),環己烷(10 mL), SOCl2(1.49 g, 12.49 mmol)和N,N-二甲基甲酰胺(DMF, 0.035 g, 0.48 mmol)),并于80 ℃條件下回流并保溫反應5 h。冷卻至室溫,減壓濃縮得到2-氟-3-三氟甲基苯甲酰氯粗品。向2-氟-3-三氟甲基苯甲酰氯粗品中加入二氯甲烷(DCM, 20 mL),冰水浴控制體系溫度不超過15 ℃,通入氨氣10 min。加入20 mL去離子水使體系中的鹽溶解,攪拌靜置分層,分出有機相。采用DCM(20 mL)萃取水相,并與有機相合并,真空濃縮得到白色固體化合物1b1.90 g,收率95.0%, m.p. 94~97 ℃;1H NMR(500 MHz, CDCl3)δ: 8.33(t,J=7.6 Hz, 1H), 7.79(t,J=7.2 Hz, 1H), 7.39(t,J=7.9 Hz, 1H), 6.63(br, 1H), 6.06(br, 1H);13C NMR(126 MHz, CDCl3)δ: 163.98, 159.15, 136.35, 130.96, 124.79, 123.38, 122.13, 121.22。
(3) 2-氟-3-三氟甲基苯胺(11)的合成
向100 mL圓底燒瓶中加入1b(2.00 g, 9.66 mmol),再加入30 mL乙醇攪拌溶解。加入質量分數為10%的NaOH溶液(4.25 g, 21 mmol),待體系溫度降至0 ℃,開始滴加濃度為1.37 mol/L NaClO溶液(7.75 mL, 10.6 mmol),滴加完畢,保溫反應40 min。升溫至80 ℃,水解脫羧,回流反應5 h。冷卻至室溫,加入50 mL去離子水使鹽溶解至出現乳化,采用乙酸乙酯(3×30 mL)萃取有機相并合并有機相,鹽水(2×30 mL)洗滌、無水Na2SO4干燥、過濾,真空濃縮得到化合物111.61 g,收率92.3%;1H NMR(500 MHz, CDCl3)δ: 7.02~6.92(m, 3H), 3.89(br, 2H);13C NMR(126 MHz, CDCl3)δ: 149.42, 135.75, 124.28, 121.92, 120.46, 118.36, 115.50。
(4) 4-溴-2-氟-3-三氟甲基苯胺(12)的合成
在100 mL圓底燒瓶中將1(2.00 g, 11.17 mmol)溶于DMF(14 mL)中,并在15 ℃條件下分次加入N-溴代琥珀酰亞胺(NBS, 1.99 g, 11.17 mmol)),保溫反應時間1 h。反應混合物用乙酸乙酯(EtOAc, 50 mL)稀釋,并用水(2×50 mL)和飽和NaCl溶液(50 mL)洗滌。分離出的有機相用Na2SO4干燥、濃縮得到化合物粗品。柱層析純化分離(洗脫劑:石油醚 ∶乙酸乙酯=10 ∶1,V∶V)得到化合物122.74 g,收率95.0%;1H NMR(500 MHz, DMSO-d6)δ: 7.31(d,J=8.7 Hz, 1H), 6.92(t,J=8.9 Hz, 1H), 5.81(s, 2H);13C NMR(126 MHz, DMSO-d6)δ: 149.41, 138.28, 131.00, 124.00, 121.82, 120.32, 102.02。
(5)N-[4-溴-2-氟-3-(三氟甲基)苯基]乙酰胺(13)的合成
向100 mL圓底燒瓶中加入化合物12(2.45 g, 9.5 mmol))和乙酸酐(6 mL)的混合物,并在25 ℃條件下攪拌反應3 h,減壓蒸餾除去剩余乙酸酐。殘留物加入冰水(10.00 g)和碳酸氫鈉(pH=7)。用乙酸乙酯(3×30 mL)萃取混合物,合并有機相,Na2SO4干燥,過濾、濃縮得到化合物132.52 g,收率88.0%, m.p.102~103 ℃;1H NMR(500 MHz, DMSO-d6)δ: 10.06(s, 1H), 8.17(t,J=8.4 Hz, 1H), 7.68(d,J=8.9 Hz, 1H), 2.12(s, 3H);13C NMR(126 MHz, DMSO-d6)δ: 169.21, 152.16, 130.71, 127.81, 127.71, 123.15, 120.98, 112.72, 23.57。
(6)N-[4-氰基-2-氟-3-(三氟甲基)苯基]乙酰胺(14)的合成
(7) 4-氨基-3-氟-2-(三氟甲基)苯甲腈(15)的合成
向100 mL圓底燒瓶中依次加入化合物14(0.70 g, 2.84 mmol)的乙醇(5 mL)溶液和濃鹽酸(12 mol/L, 5 mL)。將混合物溶液在78 ℃條件下回流反應1 h,冷卻至室溫,真空濃縮,所得固體溶于乙酸乙酯(25 mL),用飽和碳酸氫鈉溶液(25 mL)洗滌,無水MgSO4干燥,過濾、濃縮后得到淡灰色固體化合物150.54 g,收率93.0%, m.p.160~162 ℃;1H NMR(500 MHz, DMSO-d6)δ: 7.55(d,J=8.5 Hz, 1H), 7.04(t,J=8.5 Hz, 1H), 6.83(s, 2H);13C NMR(126 MHz, DMSO-d6)δ: 147.56, 142.91, 132.29, 123.01, 120.83, 118.31, 116.75, 91.94。
(8) 3-氟-4-異硫氰酸根-2-三氟甲基苯甲腈(16)的合成
在100 mL圓底燒瓶中將化合物15(0.41 g, 2 mmol)溶于四氫呋喃(THF, 5 mL),并在20 ℃條件下緩慢加入硫光氣(2 mL)水(5 mL)溶液。混合物攪拌反應1 h后濃縮。殘留物在水(50 mL)和乙酸乙酯(30 mL)之間分配。水相用乙酸乙酯(2×30 mL)萃取,合并有機相,用鹽水(2×50 mL)洗滌,無水MgSO4干燥,過濾、濃縮,得到黃色油狀液體化合物160.40 g,收率73.0%;1H NMR(500 MHz, CDCl3)δ: 7.61(d,J=8.3 Hz, 1H), 7.44(t,J=7.7 Hz, 1H);13C NMR(126 MHz, CDCl3)δ: 157.60, 146.58, 131.41, 129.50, 127.97, 121.97, 119.77, 114.40, 108.62。
2 結果與討論
2.1 合成1a的反應條件優化
合成1a的反應為鄰氟三氟甲苯的鄰位金屬化反應,通過相關文獻[10-13]發現,正丁基鋰用量、溶劑用量及反應時間對收率均存在一定影響。為了獲得較佳的反應條件,本文考察了正丁基鋰用量和反應時間對反應收率的影響。
(1) 正丁基鋰用量對1a收率的影響
以5.00 g1為原料,50 mL THF為溶劑,設置反應時間為5 h,分析正丁基鋰用量對1a收率的影響,實驗結果如表1所示。由表1可知,隨著正丁基鋰用量的加大,1a收率不斷增長。當正丁基鋰的用量為1.1 eq.時,產物1a可以獲得較佳收率(92.0%),再繼續增加正丁基鋰的用量,收率趨于穩定,變化不明顯。

表1 正丁基鋰用量對1a收率的影響
(2) 反應時間對1a收率的影響
以5.00 g1為原料,正丁基鋰用量為1.1 eq., THF為50 mL,分析反應時間對1a收率的影響,實驗結果如表2所示。由表2可知,隨著反應時間的增長,收率增大。當反應時間為5 h(95.6%)時,收率趨于穩定。

表2 反應時間對1a收率的影響
2.2 合成1b的反應條件優化
合成1b的反應為羧酸的酰氯、酰胺化反應,酰氯化指將含酰基的物質(羧基或酸酐)與含活潑氯的試劑反應,生成酰氯的過程[14]。酰氯是羧酸衍生物中最活潑的酰基化試劑,酰氯氨解可制備酰胺。據文獻[15-17]報道,影響反應收率的因素有酰氯化試劑、反應溶劑、催化劑種類和反應溫度等。本文主要考察酰氯化試劑種類和反應溶劑體系對收率的影響。
(1) 酰氯化試劑種類對1b收率的影響
以2.00 g1a為原料,酰氯化試劑均為理論量的2倍量,催化劑DMF用量為0.05 eq.,溶劑二氯甲烷為20 mL,分析不同酰氯化試劑對1b收率的影響,實驗結果如表3所示。由表3可知,當選擇氯化亞砜為酰氯化試劑時,產物1b收率最高。雖草酰氯氯化能力更強,能獲得與氯化亞砜相當的產物收率,但草酰氯毒性更大,價格昂貴,因此本文選擇氯化亞砜作為酰氯化試劑。

表3 酰氯化試劑種類對1b收率的影響
(2) 反應溶劑對1b收率的影響
以2.00 g1a為原料,氯化亞砜用量為2.0 eq.,催化劑DMF用量為0.05 eq.,氯化亞砜與溶劑的質量體積比(m/V)為1 ∶10,考察不同溶劑體系對1b收率的影響,實驗結果如表4所示。由表4可知,當溶劑為環己烷時,產物1b收率最佳(94.0%)。

表4 反應溶劑對1b收率的影響
2.3 合成11的反應條件優化
合成11的反應為Hofmann重排反應,于1881年由Hofmann[18]提出。經典反應條件以一級酰胺為起始物,在次鹵酸鹽和堿的作用下,經異氰酸酯中間體得到比起始物少1個碳原子的伯胺,所以又稱霍夫曼降解反應。霍夫曼重排反應過程中主要存在以下幾種副反應:原料酰胺的水解、產物的過度鹵化和脲及酰基脲的生成等[19]。本文考察了NaClO用量和NaOH用量對反應收率的影響。
(1) NaClO溶液用量對11收率的影響
以2.00 g1b為原料,30 mL乙醇為溶劑,NaOH用量為2.2 eq,重排反應溫度為0 ℃,重排反應時間為40 min,水解脫羧溫度為80 ℃,水解脫羧時間為5 h,分析NaClO溶液用量對11收率的影響,實驗結果如表5所示。由表5可知,當NaClO溶液用量為1.1 eq時,11收率最高(92.3%),而繼續增大NaClO溶液用量,11收率下降,該現象可能是NaClO溶液用量過多,導致產物的過度鹵化,副產物含量增大。

表5 NaClO溶液用量對11收率的影響
(2) NaOH用量對11收率的影響
以2.00 g1b為原料,30 mL乙醇為溶劑,NaClO用量為1.1 eq,重排反應溫度為0 ℃,重排時間為40 min,水解脫羧溫度為80 ℃,水解脫羧時間為5 h,考察NaOH用量對11收率的影響,實驗結果如表6所示。由表6可知,隨著NaOH用量的增大,產率先增大而后減小。當NaOH用量為2.2 eq時,11收率最高,這是由于異氰酸酯非常活潑,當增加NaOH用量時,生成的中間體異氰酸酯可以很快轉化為伯胺,但如果NaOH用量過大,N-氯代酰胺可以發生鹽析作用,導致胺化程度開始呈下降趨勢。

表6 NaOH用量對11收率的影響
本文以鄰氟三氟甲苯為原料合成關鍵前體化合物11,再經溴代、氰基取代、水解和氧化得到Proxalutamide中間體3-氟-4-異硫氰酸根-2-三氟甲基苯甲腈,并對11的合成條件進行了優化,總收率為36%。與已有合成路線相比,該合成路線的原料及試劑易得,所有反應均為常壓反應,反應條件溫和且操作簡易安全,具有較好的工業化前景。
