鈷摻雜碳活化過硫酸氫鉀降解四環素
2023-09-22 07:13:04朱紅梧汪園青向妍蕾潘育松潘成嶺
人工晶體學報 2023年9期
關鍵詞:催化劑
朱紅梧,汪園青,向妍蕾,韓 蓉,潘育松,黃 潤,杜 超,潘成嶺,2
(1.安徽理工大學材料科學與工程學院,淮南 232001;2.安徽理工大學,環境友好材料與職業健康研究院(蕪湖),蕪湖 241003)
0 引 言
近幾十年來,抗生素在醫藥、農業和畜牧業中的濫用對公共衛生和生態安全構成了重大威脅[1-2]。在水生環境中,抗生素耐藥細菌可以釋放或傳播抗生素耐藥基因,這些基因相對持久,可以被其他細菌獲得,從而產生更多的抗生素耐藥細菌。當使用抗生素時,帶有殘留抗生素的廢物是不可避免的,并最終排放到水環境中,這會破壞水生生態環境[3-4]。
四環素(tetracycline, TC)是最具代表性的抗生素之一,由于其抗菌活性,已廣泛應用于畜牧業和水產養殖等行業[5]。由于四環素優異的穩定性,其難以通過生物技術、物理吸附等傳統水處理方法去除。生物降解難以從抗生素中獲取合適的微生物[6-7],物理吸附法只能在抗生素之間積累和轉移[8-9]。而高級氧化工藝(advanced oxidation process, AOPs)已被證明是從各種超氧化物中產生高活性氧(reactive oxygen species, ROS)的強大技術,在環境修復領域越來越受到人們的關注。過硫酸鹽氧化是高級氧化技術之一,由于過硫酸鹽結構不穩定,更容易被活化產生ROS[10-11]。通過活化過硫酸鹽產生ROS來去除有機污染物的常用方法有過渡族金屬活化[12-13]、熱活化[14]、紫外線活化[15]、堿活化[16]等。過渡族金屬離子活化由于具有高效性、經濟性、便利性而成為研究熱點,其中,鈷離子被認為是活化過硫酸氫鉀(potassium monopersulfate, PMS)最有效的過渡族金屬離子之一[17]。其均相催化雖然具有很高的活性和選擇性,但難以從反應體系中分離出來,不能重復使用,甚至可能給環境帶來重金屬離子的二次污染,不利于工業應用。為了克服均相催化劑的缺點,研究人員將均相催化劑固載化制備非均相催化劑[18]。非均相催化劑不僅可以很好地解決問題,還賦予了催化劑特定的性能[19]。碳組分通常可以作為輔助提高金屬性能的添加劑與載體、金屬離子與導電碳納米材料(如石墨烯[20]、碳納米管[21]等)的復合,增強金屬/碳復合材料的電荷轉移能力,也為增強催化材料的化學性能和結構穩定性提供了有效方法。但不同金屬/碳復合雜化材料仍然存在著制備工序復雜和成本高的問題。其次,金屬離子摻雜碳球屬于一個新的研究領域,有關報道較少,且方法不具普適性,缺乏利用金屬離子定制碳球的新性能與新應用。
本文采用簡易的水熱法與原位負載相結合,探究Co/C/PMS體系對TC的降解能力,不僅考察了鈷摻雜量對催化劑降解TC的影響,還研究了其他因素(催化劑投加量、PMS的投加量、pH、無機陰離子)對TC降解效率的影響,并進一步結合自由基分析,探討Co/C/PMS體系降解TC的作用機理。
1 實 驗
1.1 實驗原料
無水葡萄糖(純度99%)、六水合硝酸鈷(純度99%)、過硫酸氫鉀、氯化鈉(純度99.5%)、叔丁醇(純度99.5%)、L-組氨酸(純度99%)、對苯醌(純度99%)均購于上海麥克林試劑廠,四環素(純度99%)、氫氧化鈉(純度99%)、無水硫酸鈉(純度99%)、無水碳酸鈉(純度99%)、無水乙醇(純度99.5%)均購于阿拉丁試劑廠,甲醇購于上海邁瑞爾化學試劑廠。實驗中所有用水均為超純水。
1.2 制備方法
采用水熱合成與原位負載相結合的方式制備鈷負載碳基催化材料。具體制備工藝如下:
稱取3份0.026 mol的無水葡萄糖粉末分別倒入3個裝有50 mL去離子水的燒杯中并充分溶解。再稱取0.005 mol、0.01 mol、0.015 mol六水合硝酸鈷分別緩慢加入葡萄糖溶液中,以800 r/min攪拌4 h使其充分溶解后,將混合溶液分別倒入100 mL的高壓反應釜中于180 ℃下保溫12 h。隨后使用去離子水和無水乙醇反復清洗產物至少6次,最后將產物離心過濾后置入60 ℃的烘箱中充分干燥,將干燥后得到的黑色產物依次記作Co/C-1、Co/C-2、Co/C-3。此外,相同步驟合成了不摻鈷的純水熱碳,并將棕褐色產物記作C。
1.3 表 征
采用D8 Avance型X射線衍射儀對樣品進行XRD表征。將粉末樣品在樣品板的凹槽內壓實。掃描速率為5 (°)/min,掃描角度范圍為10°~80°,電壓40 kV,電流30 mA。
采用低電壓冷場SU8100型掃描電子顯微鏡對樣品進行SEM表征。將少量樣品加入無水乙醇中超聲分散后制樣并噴金處理,觀察樣品的微觀結構與形貌。
采用美國FEI-TecnaiG2型透射電子顯微鏡對樣品進行TEM表征。將少量樣品加入無水乙醇中超聲分散后制樣并噴金處理,觀察樣品的微觀結構與形貌。
采用型號為美國Thermo Scientific ESCALAB Xi+的X射線光電子能譜儀對樣品進行XPS分析,檢測了樣品表面元素及其化學狀態。
1.4 性能測試
配制濃度為20 mg/L的TC溶液,使用紫外分光光度計在357 nm的最大吸收峰處測定吸光度。
將50 mL濃度為20 mg/L的TC溶液置于燒杯中,向燒杯中加入催化劑樣品。在黑暗環境下吸附60 min,吸附達到平衡后加入250 μL濃度為0.1 mol/L的PMS并開始計時,分別在0、10、20、30、40、50、60 min取1 mL溶液立刻倒入裝有1 mL甲醇的管中淬滅,使用0.22 μm水系微孔膜過濾。測定TC在不同時間的吸光度,并通過其標準曲線計算降解率。
降解率計算公式為
(1)
式中:η為降解率,C0為溶液中TC的初始濃度,Ct為t時刻TC的濃度。
2 結果與討論
2.1 催化劑的表征與分析
2.1.1 XRD分析
通過圖1所示的XRD圖譜可以看到,葡萄糖水熱碳化產物C在22 °處具有明顯的寬衍射特征峰,與石墨碳結構面間距(002)相對應[22]。此外,Co/C產物在22°處均出現明顯寬的衍射特征峰,表明鈷離子摻雜對石墨碳結構沒有影響,但是可能因反應溫度并不高而沒有發現碳鈷化合物及氧化鈷的特征峰,這也預示著Co/C產物中摻雜的鈷離子的存在形式為離子或單質狀態。Co/C產物衍射峰強度更高,這表明產物晶相含量大,Co/C產物較水熱碳化產物C進一步聚合[23]。
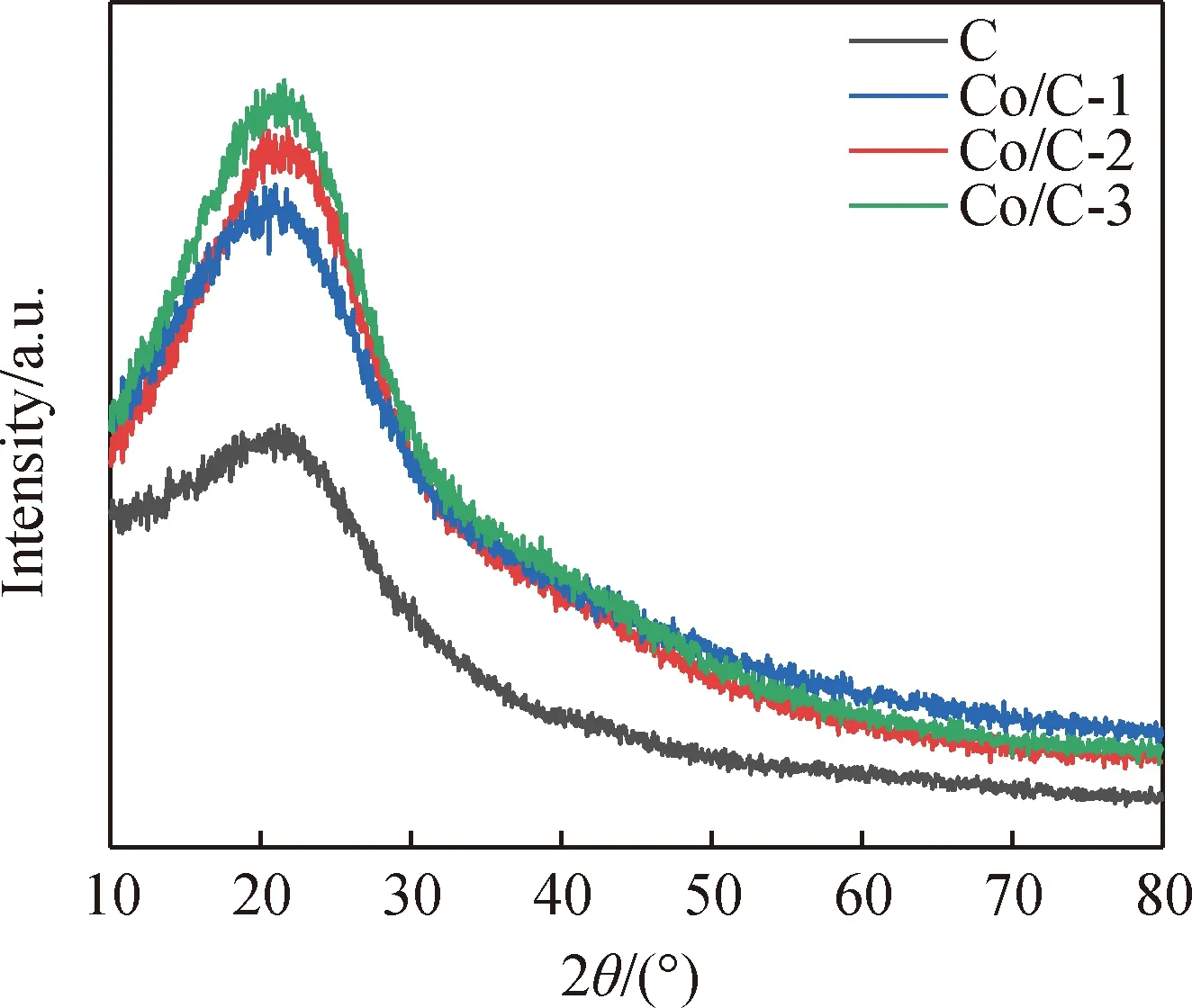
圖1 C及Co/C的XRD圖譜Fig.1 XRD patterns of C and Co/C
2.1.2 SEM表征
圖2為C和Co/C的SEM照片,通過照片可以清晰看到C和Co/C產物的微觀形貌。圖2(a)和圖2(b)中觀察到水熱碳結構是納米級的粒子聚集組成微米級的粘連球狀,通過高倍SEM照片不僅可以更加清晰地看出碳球的粘連狀態,還可以發現水熱制備的碳球直徑為300~350 nm。這種粘連狀態可能是大部分粒子由于溶劑蒸發緩慢,初級粒子融合及分級結構界限不明顯,從而碳微球由粒徑極小的納米粒子緊密堆積組裝而成[24]。圖2(c)和圖2(d)為Co/C-2的SEM照片,在形貌上仍為幾何結構穩定的球狀,但在分散度上與碳球有著很大的差別,經過鈷離子摻雜后的碳球明顯變大,球體直徑為5~10 μm。顆粒粒徑增大的原因是鈷離子的引入改變了水熱過程中碳球的移動,粒子間擴散能力增強,粒徑之間吞并的速率加劇,故而會出現碳球變大現象。相比之下,摻雜鈷離子后碳球表面缺陷明顯增多,造成這種現象的原因可能是鈷離子摻雜在一定程度上可以修改原始葡萄糖碳化成球的形態特征,且水熱過程中伴隨著碳球表面電負性的改變,使得微觀下的碳球粗糙且具有缺陷[24-25]。圖2(e)和圖2(f)分別為Co/C-1和Co/C-3的SEM照片,過量的鈷離子會使得碳球缺陷增多,對性能也會有很大的影響。

圖2 C及C/Co的SEM照片。(a)、(b)C的SEM照片;(c)、(d)Co/C-2的SEM照片;(e)Co/C-1的SEM照片;(f)Co/C-3的SEM照片Fig.2 SEM images of C and C/Co. (a), (b) SEM images of C; (c), (d) SEM images of Co/C-2; (e) SEM image of Co/C-1; (f) SEM images of Co/C-3
為了進一步表征材料內部結構和碳和鈷元素,Co/C-2的EDS展示出了碳和鈷的元素分布,鈷元素分布圖與碳基體重疊,且元素均勻地分布在碳基結構和邊緣,這表明鈷元素均勻地摻雜進碳基質微球中。如圖3(d)所示,Co/C-2表面主要由C和Co元素組成,質量分數分別為99.27%和0.73%,原子數分數為99.85%和0.15%,再次證實了鈷元素成功摻雜到碳球中。
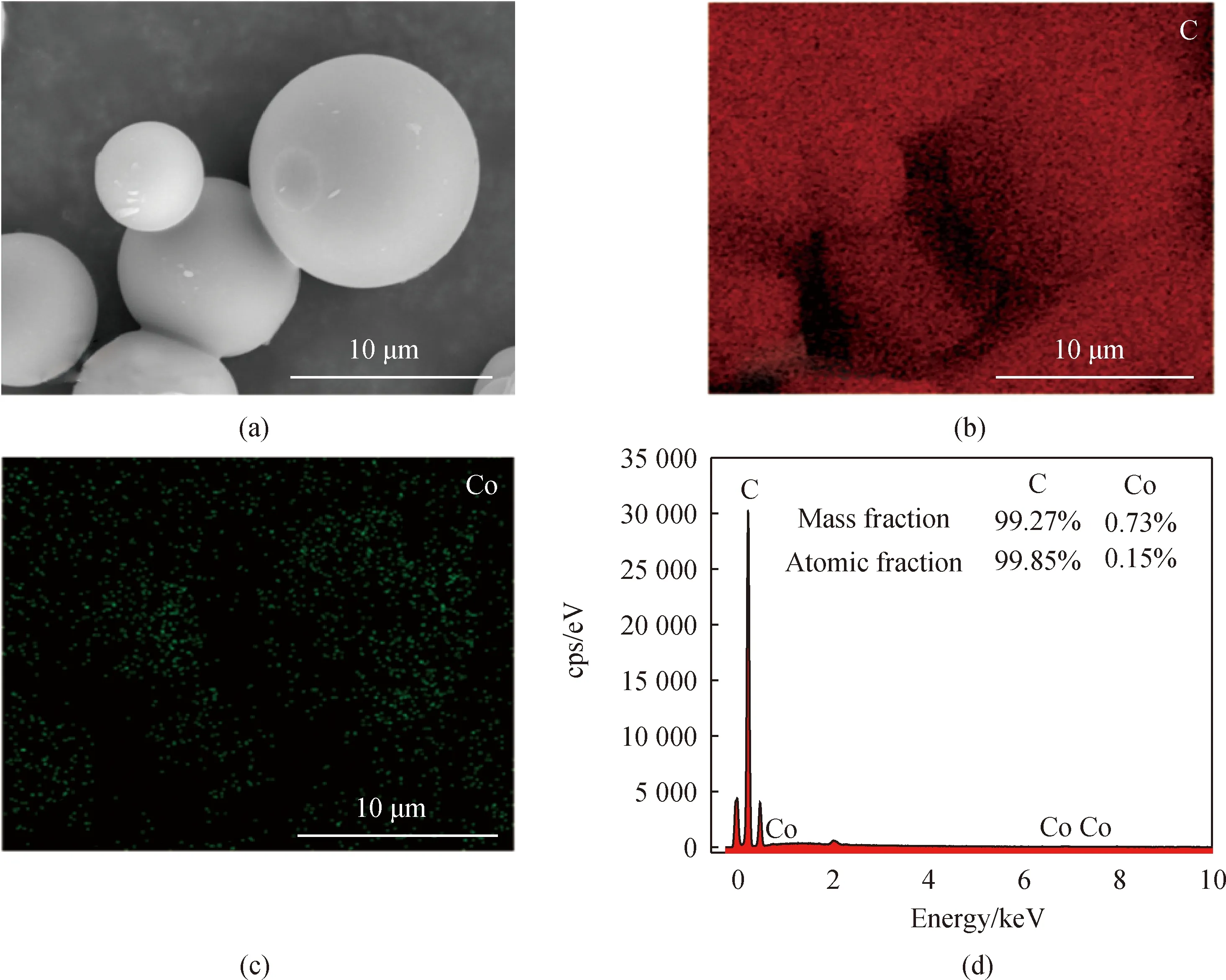
圖3 Co/C-2的EDS圖及元素含量分布圖。(a)Co/C-2的EDS面掃描圖;(b)碳元素分布圖;(c)鈷元素分布圖;(d)元素含量分布圖Fig.3 EDS element surface scans and element content distribution maps of Co/C-2. (a) EDS elemental surface scan map of Co/C-2; (b) carbon distribution map; (c) cobalt distribution map; (d) element content distribution map
2.1.3 TEM表征
Co/C-2的TEM照片如圖4所示,可以看出Co/C-2呈現均勻分布的球形納米粒子結構,并具有明顯的晶格結構,通過高分辨率電鏡可以發現其晶格間距為0.18 nm,接近于鈷單質的(200)晶面,對應的鈷納米顆粒直徑大約在7 nm[26]。同時在金屬鈷單質的周圍觀察到明顯的石墨層,條紋間距約為0.35 nm[27]。這些結果進一步證實了鈷納米顆粒的存在,并且鈷納米顆粒很好地陷入了碳基質中。這可能是鈷離子與葡萄糖水熱反應時被葡萄糖還原成了部分鈷單質,未還原部分仍依舊以離子的形式存在于碳基質中。然而當摻入鈷元素后,由于Co2+的靜電排斥作用很大程度上提高了碳球之間的分散度,能夠使其經歷溫和碳化過程形成尺寸均一的Co/C-2。
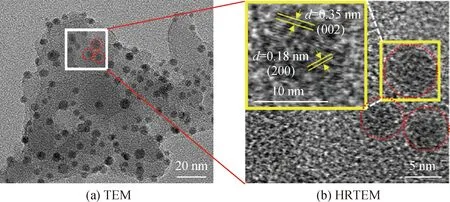
圖4 Co/C-2的TEM照片Fig.4 TEM images of Co/C-2
2.1.4 XPS分析


圖5 C及Co/C-2的XPS總譜圖及分譜。(a)C及Co/C-2的XPS圖;(b)水熱碳C 1s圖譜;(c)Co/C-2的C 1s圖譜;(d)反應前Co 2p圖譜;(d)反應后Co 2p圖譜Fig.5 XPS total spectra and spectral analysis of C and Co/C-2. (a) XPS of C and Co/C-2; (b) C 1s spectra of hydrothermal carbon; (c) C 1s spectra of Co/C-2; (d) Co 2p spectra before reaction; (e) Co 2p spectra after reaction
2.2 催化劑活化PMS降解TC的影響
2.2.1 不同催化劑活化PMS對TC降解的影響
在室溫環境下首先測試由不同配比合成催化劑的催化性能。如圖6(a)所示,所有催化劑在暗反應60 min后達到吸附平衡且物理吸附作用都很低,表明該催化劑物理吸附對TC的去除沒有顯著影響。PMS加入反應60 min后,C、Co/C-1、Co/C-2、Co/C-3對TC的降解效率分別為28.39%、59.40%、89.50%、70.00%。黃曉丹等[32]通過制備Co3O4@C降解羅丹明6G,當溶液初始pH為6.5、Co3O4@C投加量為0.1 g/L、PMS濃度為0.5 mmol/L時,反應120 min后5 mg/L羅丹明6G的降解率才達96.1%。相比之下,該材料性能具有一定的優異性,所制備的鈷摻雜水熱碳材料的催化活性能夠得到顯著提高。這可能歸因于鈷離子活化PMS而產生活性自由基,實現污染物快速降解[33-34]。
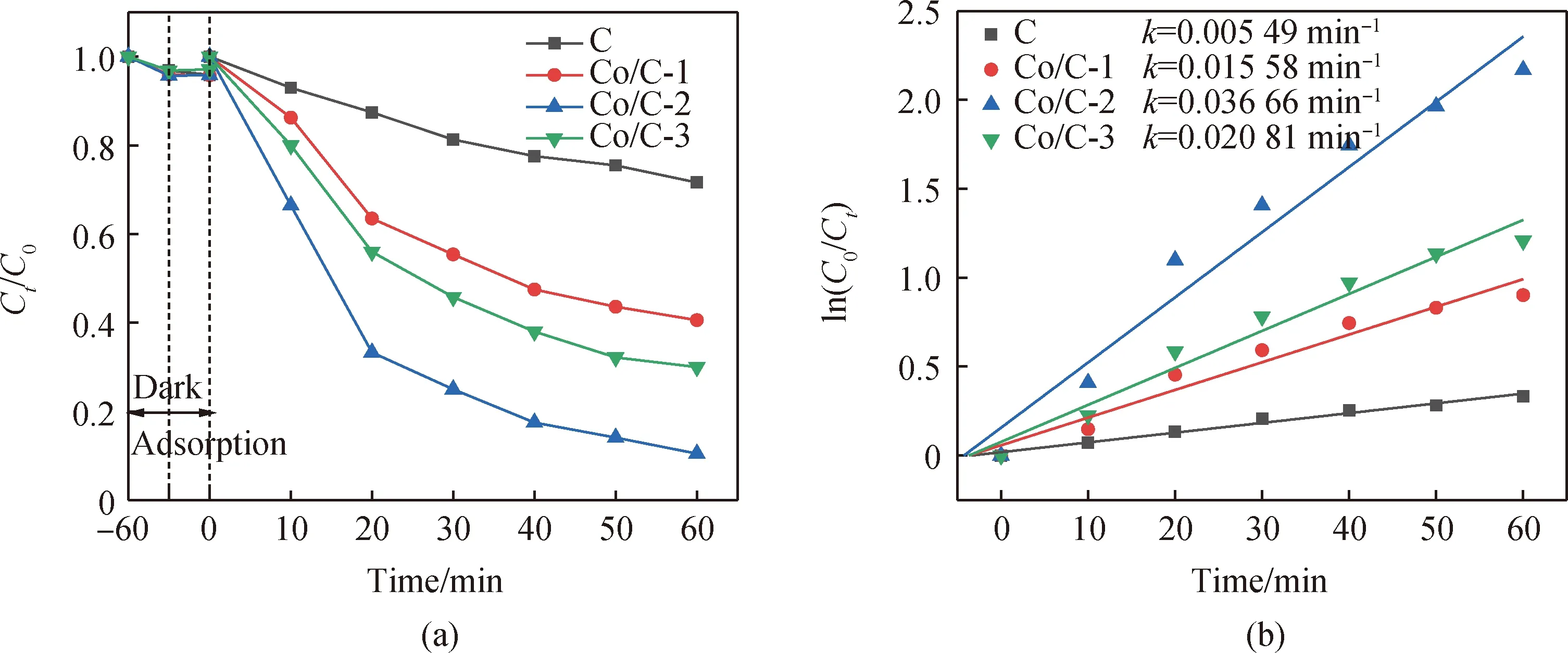
圖6 不同催化劑對TC降解(a)及TC催化表觀速率(b)的影響(實驗條件:50 mL濃度為20 mg/L的TC溶液,催化劑投加量20 mg,PMS投加量250 μL,pH=5.5)Fig.6 Effect of different catalysts on TC degradation (a) and apparent rate (b) for TC (experimental conditions: 50 mL of 20 mg/L TC solution, catalyst dosage of 20 mg, and PMS dosage of 250 μL, pH=5.5)
根據一級動力學方程式(2)計算反應速率常數k。
(2)
Co/C-2的反應速率常數分別是C、Co/C-1、Co/C-3的6.67倍、2.35倍、1.76倍。由此可見并不是摻的鈷離子越多性能越好,可能原因是:在低濃度下增加鈷的含量會導致其與PMS發生碰撞,從而促進活性自由基的產生,但在達到一定濃度后,鈷元素濃度已滿足Co0、Co2+和Co3+的循環轉換[35],所以繼續增加六水合硝酸鈷用量時TC的降解率沒有明顯變化;鈷離子摻雜在一定程度上可以修改原始葡萄糖碳化成球的形態特征,且水熱過程中伴隨著碳球表面電負性的改變,使得微觀下的碳球粗糙且具有缺陷,并又出現了融合的球體和不規則的顆粒,對性能也會有很大的影響。因此,合成催化劑的原材料偶聯也可以有效提高催化性能。
2.2.2 Co/C-2投加量的影響
如圖7所示,本文研究了Co/C-2催化劑投加量的影響。當Co/C-2催化劑的投加量從10 mg增加到40 mg,暗吸附產生的物理降解有所增加,TC的降解率也逐漸增加。當Co/C-2投加量在40 mg時,60 min內TC的降解率可以達到95.84%,這說明Co/C-2催化劑投加量的增大有利于提供更充足的反應活性位點,促進Co/C-2對PMS的活化作用,從而產生更多的氧化體系,加速降解反應的進行[36]。當催化劑用量逐漸增加時,逐漸增強的催化作用還可以判斷為由PMS消耗所引起的,故單一增加催化劑用量并不是提高TC的降解率的唯一途徑。
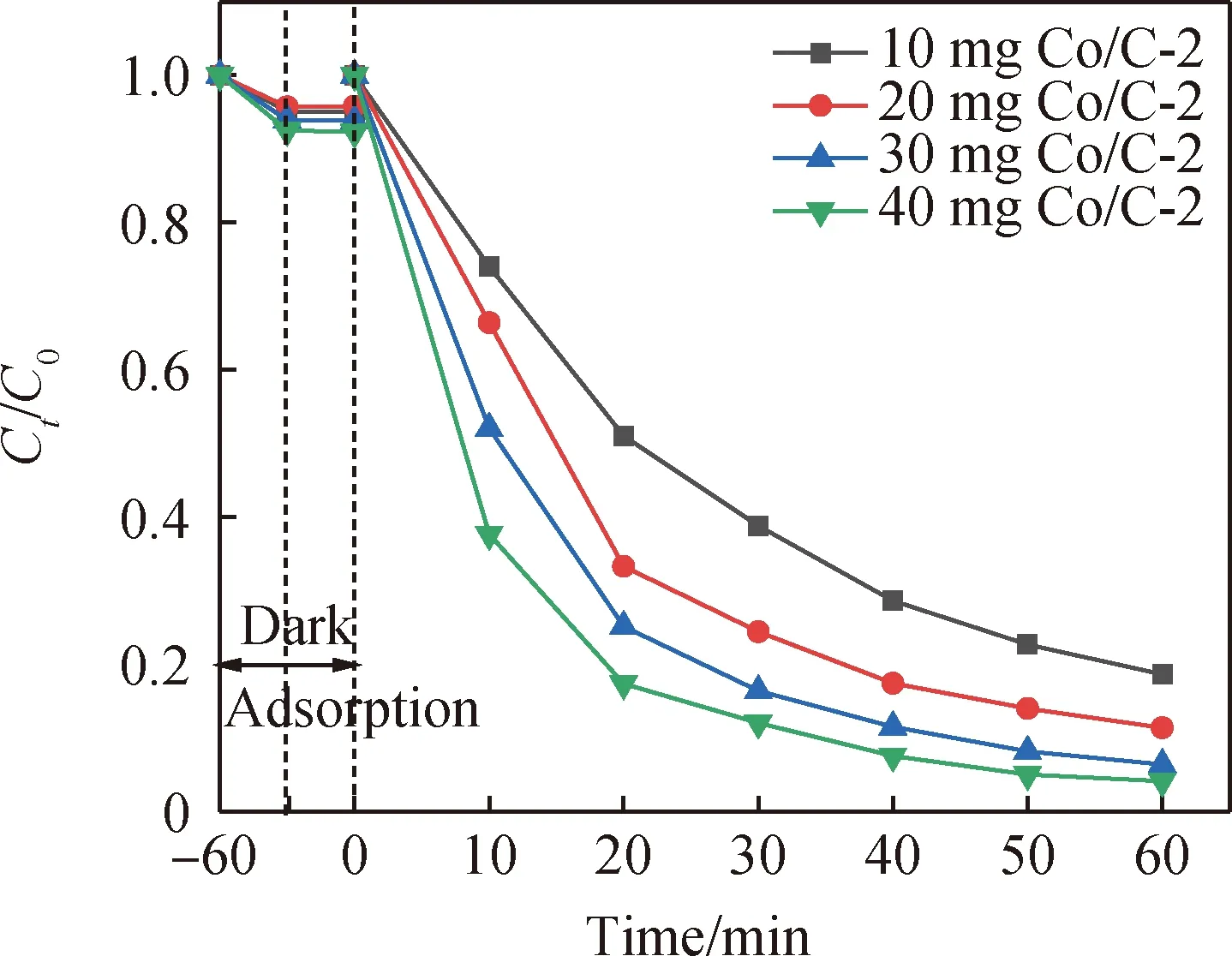
圖7 Co/C-2投加量對TC降解的影響(實驗條件:50 mL濃度為20 mg/L的TC溶液,PMS投加量為250 μL,pH=5.5)Fig.7 Effect of Co/C-2 dosage on TC degradation (experimental conditions: 50 mL of 20 mg/L TC solution, PMS dosage of 250 μL, pH=5.5)
2.2.3 PMS投加量的影響
如圖8所示,本文研究了PMS投加量對Co/C-2降解TC的影響。隨著PMS投加量的進一步增大,體系中的TC降解速率也逐漸增大。當PMS投加量增大至1 mL時,60 min內對TC的降解率可達到98.99%(k=0.086 67 min-1),反應速率常數是PMS投加量為150、250、500 μL時的2.71、1.69、1.51倍。這是因為當Co/C-2/PMS體系中的PMS投加量增大,其與催化劑表面的活性位點相接觸并發生氧化反應的概率增大,從而增大了反應體系中活性自由基的濃度,進而提高了TC被降解的速率。但當PMS投加量為500 μL和1 mL時,盡管反應速率常數k值高于250 μL的體系,但PMS投加量的增大也會造成水體中污染離子濃度的增大,從而造成水體的二次污染[37]。而催化劑不溶于TC污染物溶液,通過簡單的過濾即可去除。因此,綜合對TC的降解率、經濟成本及環境等因素考慮,Co/C-2催化劑和PMS最佳的投加量分別為40 mg和250 μL。
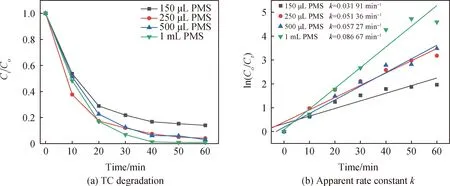
圖8 PMS投加量對TC降解及催化表觀速率的影響(實驗條件:50 mL濃度為20 mg/L的TC溶液,Co/C-2投加量為40 mg,pH=5.5)Fig.8 Effect of PMS dosage on TC degradation and apparent rate (experimental conditions: 50 mL of 20 mg/L TC solution, 40 mg of Co/C-2, pH=5.5)
2.2.4 不同陰離子的影響
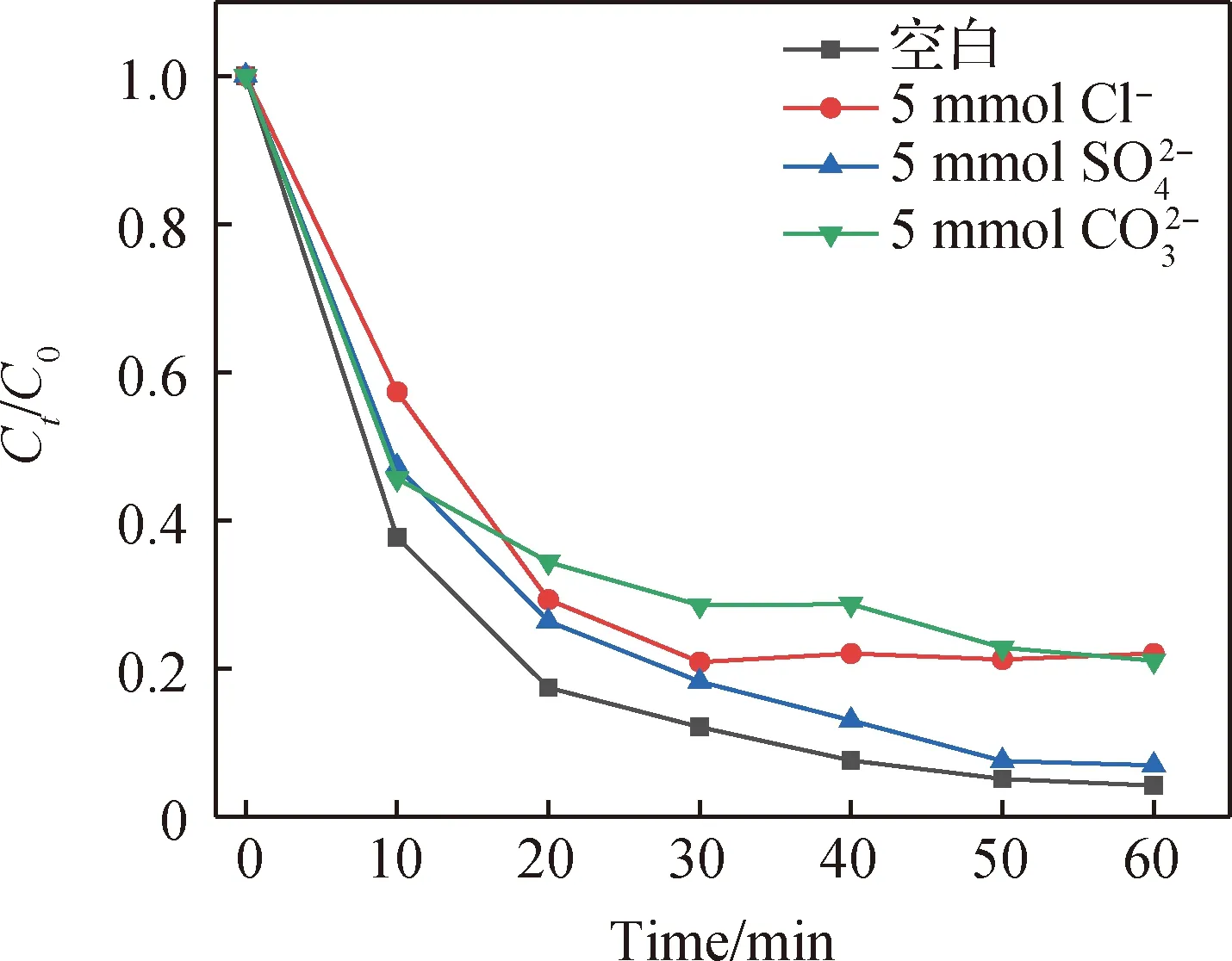
圖9 無機陰離子對TC降解的影響(實驗條件:50 mL濃度為20 mg/L的TC溶液,Co/C-2投加量為40 mg,PMS投加量為250 μL,pH=5.5)Fig.9 Effect of inorganic anions on TC degradation (experimental conditions: 50 mL of 20 mg/L TC solution, 40 mg of Co/C-2, PMS dosage of 250 μL, pH=5.5)
(3)
Cl-+·OH→ClOH·-
(4)
(5)
2.2.5 不同pH值的影響
溶液的pH值會影響碳球表面電負性和Co/C-2與PMS之間的反應,其是影響Co/C-2活化PMS降解TC的重要因素之一。如圖10所示,考察了初始pH值對TC降解效率的影響。實驗中分別使用0.1 mol/L H2SO4和0.1 mol/L NaOH調節TC溶液的初始pH值為2.5和11。結果顯示,在較寬的pH值范圍(2.5~11),經60 min反應后,TC的降解率均可在80%以上,說明Co/C-2/PMS體系在寬的pH值范圍內仍可以保持較高催化活性,但在不同pH值條件下存在差異。當pH值由5.5降到2.5時,可以發現Co/C-2/PMS體系對TC的降解率稍有下降。這是因為酸性環境下,抑制TC降解的原因可能是:PMS會與H+結合形成氫鍵,阻礙了PMS與Co/C-2間的相互作用;H+會與產生的活性物種反應,從而減弱TC的降解效果[42];此外,PMS在酸性環境下更穩定,不易產生活性物種。當pH值為5.5時,TC與Co/C-2之間的靜電斥力減小,H+抑制作用減弱,TC的降解率增高。當pH值由5.5上升至11時,可以發現Co/C-2/PMS體系對TC的降解效率在30 min前略有上升,而之后降解速率緩慢下降。堿性條件下前期的TC降解速率顯著增加的主要原因可能是:在堿性溶液中,TC主要以去質子化狀態存在,這也使得其更易被自由基氧化[43];PMS在堿性條件下也容易分解形成1O2,過量的OH-提供更多的電子以促進催化反應。而30 min后,Co2+發生沉淀,逐漸減弱了催化劑活化PMS的能力,因此TC的降解率和反應速率減小。

圖10 pH對TC降解的影響(實驗條件:50 mL濃度為20 mg/L的TC溶液,Co/C-2投加量為40 mg,PMS投加量為250 μL)Fig.10 Effect of pH on TC degradation (experimental conditions: 50 mL of 20 mg/L TC solution, 40 mg of Co/C-2, PMS dosage of 250 μL)
2.2.6 Co/C-2的循環性能分析
循環性能是催化劑性能的一個重要指標,因而對Co/C-2催化劑的循環性能進行了測試,結果如圖11所示,經2次和3次循環后,四環素的降解率分別為87.17%和75%。結果表明該材料具有一定的重復利用性,但是不適合多次循環利用。通常情況下含金屬催化劑對PMS的活化均會表現出較差的循環性能,因為部分金屬離子在反應過程中發生了不可逆轉的氧化還原反應,或者可能是材料表面的孔道被阻塞,活性位點的喪失,電子傳遞受到了阻礙,造成了催化劑失活。相比之下,碳基催化劑對PMS活化表現出較差的循環性能通常與其表面化學和結構的改變有關[44-45]。

圖11 Co/C-2的循環性能圖(實驗條件:50 mL濃度為20 mg/L的TC溶液,Co/C-2投加量為40 mg,PMS投加量為250 μL,pH=5.5)Fig.11 Cycle performance graph of Co/C-2 (experimental conditions: 50 mL of 20 mg/L TC solution, 40 mg of Co/C-2, PMS dosage of 250 μL, pH=5.5)
2.2.7 活性物種分析

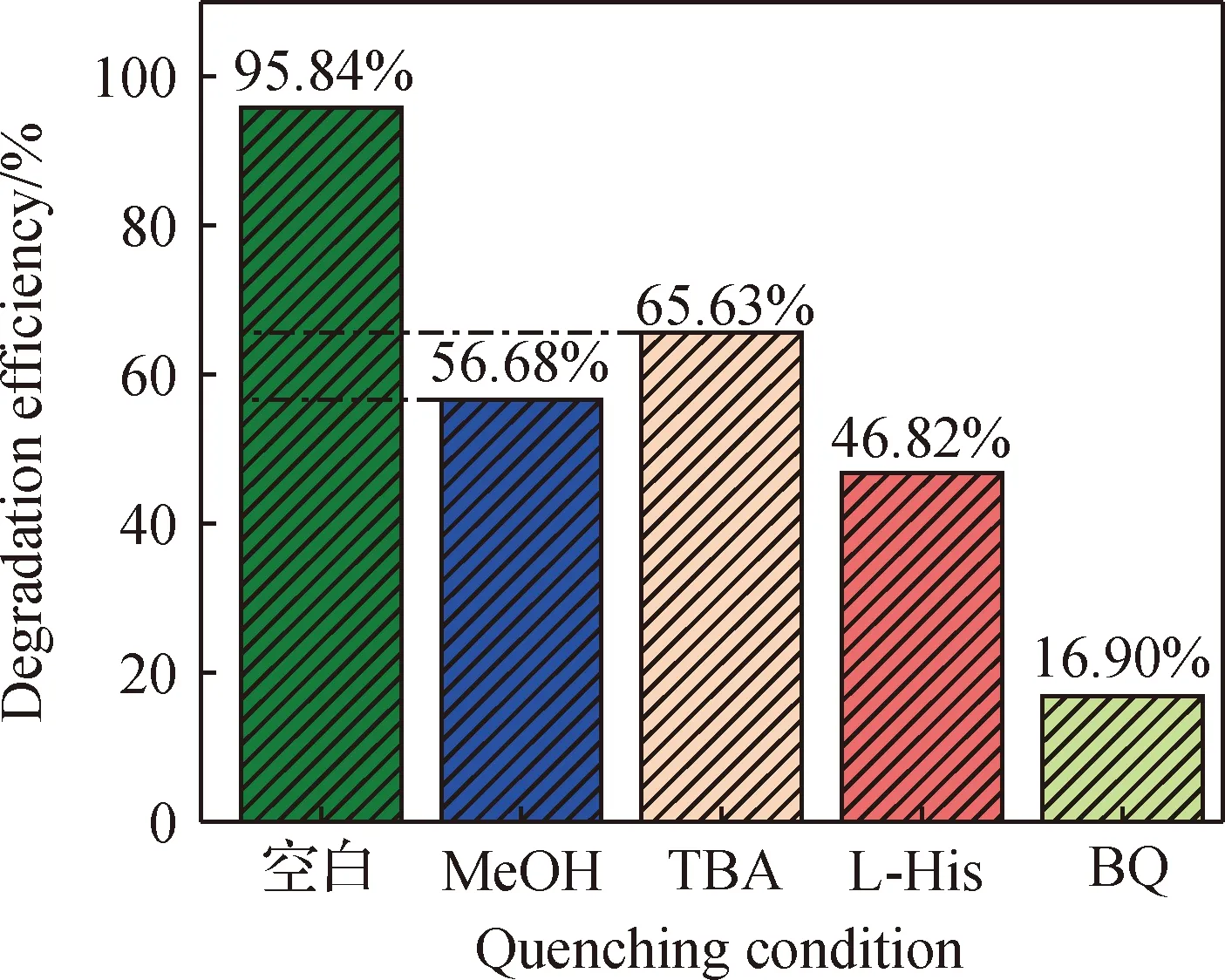
圖12 捕獲劑對Co/C-2/PMS體系降解TC的影響(實驗條件:50 mL濃度為20 mg/L的TC溶液,Co/C-2投加量為40 mg,PMS投加量為250 μL,pH=5.5)Fig.12 Effect of capture agent on degradation of TC in Co/C-2/PMS system (experimental conditions: 50 mL of 20 mg/L TC solution, 40 mg of Co/C-2, PMS dosage of 250 μL, pH=5.5)
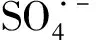
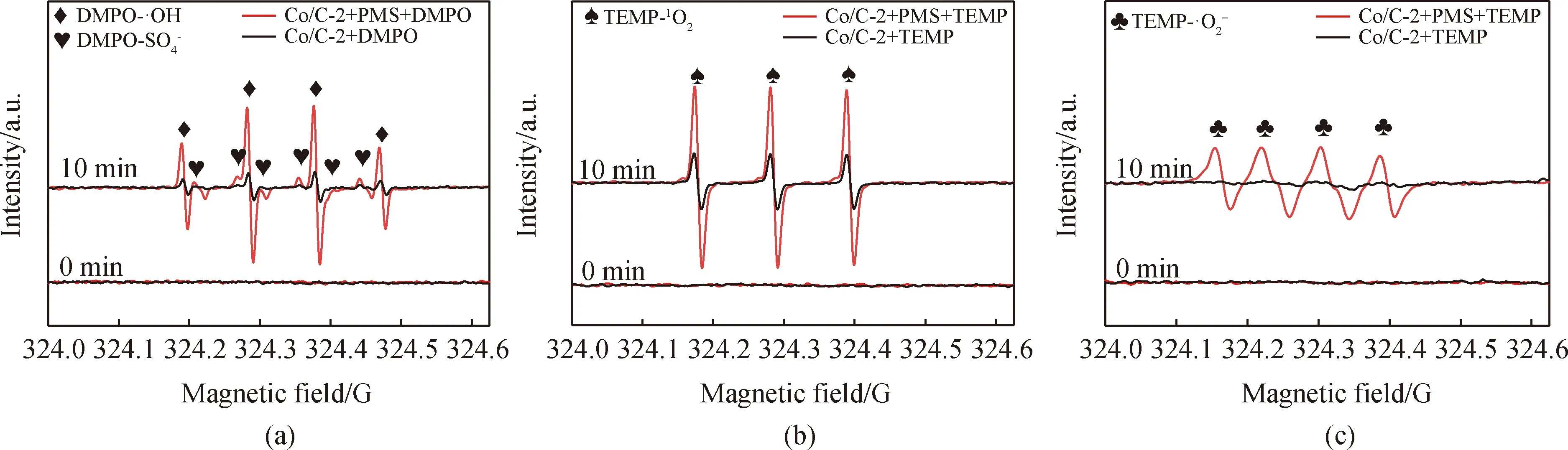
圖13 Co/C-2/PMS體系的EPR圖譜。(a)Co/C-2/PMS/DMPO的EPR圖譜;(b)Co/C-2/PMS/TEMP-1O2的EPR圖譜;的EPR圖譜Fig.13 EPR maps of Co/C-2/PMS system. (a) EPR map of Co/C-2/PMS/DMPO; (b) EPR map of Co/C-2/PMS/TEMP-1O2; (c) EPR map of
2.2.8 Co/C-2活化PMS機理分析及可能產生的中間體

圖14 Co/C-2激活PMS降解TC機理圖Fig.14 Mechanism diagram of TC degradation by Co/C-2 activated PMS
(6)
(7)
(8)
(9)
(10)
(11)
(12)
(13)
(14)
(15)
(16)
3 結 論
通過簡單的水熱法與原位負載相結合制備了摻鈷碳基化合物催化劑。采用Co/C/PMS體系降解TC,分析了各個降解體系的降解能力,還考察了各因素對TC降解過程的影響,并通過捕獲實驗和EPR測試確定了反應自由基。主要結論如下:
1)不同摩爾比的Co/C材料在降解TC存在差異,其中,Co/C-2材料具有最大的降解率和最快的降解速率,且降解效果遠遠優于單一的水熱碳。
2)Co/C-2/PMS體系可以有效降解TC,在最佳的條件下(Co/C-2投加量為40 mg,PMS投加量為250 μL,pH=5.5,反應時間為60 min)下,TC的降解率為95.84%(k=0.051 36 min-1)。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
