分散固相萃取結(jié)合UPLC-MS/MS測定不同人參加工品中46 種皂苷類化合物
2023-10-21 03:14:46王艷紅吳雨桐曹虹芳許煊煒李月茹
食品科學 2023年18期
關(guān)鍵詞:檢測
王艷紅,吳雨桐,曹虹芳,許煊煒,趙 丹,李月茹,*
(1.吉林農(nóng)業(yè)大學 農(nóng)業(yè)農(nóng)村部參茸產(chǎn)品質(zhì)量監(jiān)督檢驗測試中心,吉林 長春 130118;2.貴州中醫(yī)藥大學藥學院,貴州 貴陽 550025;3.吉林農(nóng)業(yè)大學中藥材學院,吉林 長春 130118)
人參是五加科植物人參(Panax ginsengC.A.Meyer)的干燥根和根莖,具有大補元氣、復脈固脫、補脾益腎、生津養(yǎng)血、安神益智之功效[1],是我國傳統(tǒng)的名貴中藥材,一直以來素有“中藥之首,百草之王”的美譽。現(xiàn)代藥物化學和藥理學研究結(jié)果表明,人參中皂苷類物質(zhì)是其主要的活性成分,具有抗腫瘤、抗衰老、抗氧化、降血糖和增強免疫功能等多種藥理作用[2-3]。人參皂苷根據(jù)在人參中的量被分為常量人參皂苷(主要人參皂苷)和稀有人參皂苷,常量人參皂苷如人參皂苷Ra1、Ra2、Ra3、Rb1、Rb2、Rb3、Rc、Rd、Re、Rg1、Rg2、Rf等含有較多糖基、在人參中含量相對較高,但其生物活性較低,在人體內(nèi)的吸收率也很低;稀有人參皂苷如人參皂苷Rg3、Rg5、Rg6、F4、Rh1、Rh2、Rh3、Rh4、Rk1、Rk2、Rk3、F1、F2等在人參屬植物中含量很少,很難從人參屬植物中直接分離得到,一般通過降解常量人參皂苷的方法制備稀有人參皂苷,但稀有人參皂苷含糖基較少,生物活性更好,身體吸收率更高[4-5]。此外,人參通過不同的加工方式,部分常量皂苷也會發(fā)生轉(zhuǎn)化,產(chǎn)生稀有人參皂苷[6-7]。市場上最常見的人參加工產(chǎn)品為生曬參和紅參,近年來,人參經(jīng)過九蒸九曝加工方法而成的黑參也逐漸出現(xiàn)在市場上,且由于黑參除了部分稀有皂苷含量較高,還含有一些特異性的稀有人參皂苷,并表現(xiàn)出更強的生物活性,越來越受到廣大消費者的歡迎[8]。可見不同的加工方式引起了人參皂苷含量和種類的差異,對其藥理活性和臨床應用也有較大的影響。為此,評估人參皂苷水平對人參加工產(chǎn)品的質(zhì)量控制和正確有效使用至關(guān)重要。而人參皂苷種類繁多,且結(jié)構(gòu)具有多樣性和相似性,針對人參相關(guān)產(chǎn)品復雜體系中多種皂苷類成分的快速鑒定及定量分析檢測技術(shù),國內(nèi)外研究者們也一直在不斷的探索和研究。
近20年來,利用高效液相色譜法和超高效液相色譜法定量分析人參皂苷的含量已經(jīng)是一種比較成熟的分析手段[9-12],但由于檢測種類有限,分析時間較長,單一液相色譜方法難以滿足人參及人參相關(guān)產(chǎn)品復雜體系中多種皂苷類成分同時快速分析檢測。隨著現(xiàn)代質(zhì)譜技術(shù)的快速發(fā)展,液相色譜-質(zhì)譜聯(lián)用技術(shù)因其具有靈敏度高、分辨率好且高效快速便捷等優(yōu)點已被廣泛用于人參皂苷定性定量分析研究中。目前,雖然已有大量研究報道了應用液相色譜-質(zhì)譜聯(lián)用技術(shù)分析及鑒定人參皂苷類成分的檢測方法,從液相色譜-質(zhì)譜聯(lián)用技術(shù),到超高效液相色譜-高分辨質(zhì)譜聯(lián)用技術(shù)等。但既簡單、準確、快速又覆蓋皂苷種類多的定量檢測分析方法還有限。三重四極桿質(zhì)譜通常采用的多反應監(jiān)測(multiple reaction monitoring,MRM)模式需要已知的標準品優(yōu)化儀器參數(shù)和對分析物進行定量分析檢測,具靈敏度高、選擇性強的優(yōu)勢,更適用于已知物質(zhì)的定量分析。如Xia Yonggang等[13]采用超高效液相色譜結(jié)合電噴霧電離和三重四極桿質(zhì)譜分析測定了西洋參中22 種人參皂苷的含量;石婧婧等[14]建立了超快速液相色譜-三重四極桿/線性離子阱質(zhì)譜法同時測定紅參中17 種人參皂苷類成分;趙立春等[15]建立了人參中28 種人參皂苷含量的超高效液相色譜-質(zhì)譜法(ultra-high performance liquid chromatography-tandem mass spectrometry,UPLC-MS/MS)檢測方法。這些超高效液相色譜-三重四極桿質(zhì)譜法,操作簡單、快速準確,但是分析皂苷種類和數(shù)量覆蓋面較窄,難以滿足人參不同產(chǎn)品中皂苷類化合物復雜多樣的分析檢測要求。郝穎[16]和戴雨霖[17]等采用了高分離度快速液相色譜-四極桿-飛行時間質(zhì)譜法分析鑒定了不同人參樣品中40多種皂苷類成分;Lee等[18]采用超高效液相色譜-高分辨飛行時間質(zhì)譜聯(lián)用技術(shù)分析鑒定了人參根、莖、葉和果中的58 種皂苷類化合物。這些高分辨質(zhì)譜法檢測人參皂苷的多組分常采用飛行時間質(zhì)譜檢測器,具有超高的分辨率和精確分子質(zhì)量測定功能,更適用于未知物質(zhì)的定性鑒別。且超高效液相色譜-高分辨質(zhì)譜聯(lián)用儀不僅價格昂貴,還需具備較高的專業(yè)技術(shù)水平等,在大部分領(lǐng)域的應用仍處于發(fā)展階段,大部分實驗室仍未普及,液相色譜-三重四極桿串聯(lián)質(zhì)譜儀仍然是當前科研和檢測機構(gòu)的主流定量分析手段。
由于人參不同產(chǎn)品中含有較多的糖類、脂類及色素等成分,為了降低基質(zhì)的干擾,保證數(shù)據(jù)的準確性,并減少對色譜柱和儀器的污染,需要對其進行凈化處理。目前最常見的人參皂苷前處理凈化方法仍是傳統(tǒng)的固相萃取法[19-20]、液液萃取法[21-22],這些方法存在過程繁瑣、耗時長、成本高、需要大量有毒有機溶劑等問題,且無法滿足批量樣品中多種皂苷的同時簡單、快速、準確檢測的需求。分散固相萃取法作為一種簡便、快速、價格低廉的分析方法,已成功用于農(nóng)藥殘留檢測領(lǐng)域。近年來該方法在中藥有效成分提取凈化及分析中的應用研究也有報道,如藥用狗牙花中生物堿、五味子中木脂素[23-24]。但將分散固相萃取法用于人參中皂苷類成分凈化分析方面的研究鮮見報道。本研究在前人研究基礎(chǔ)上,采用了分散固相萃取法對人參樣品進行凈化處理,利用超高效液相色譜-三重四極桿串聯(lián)質(zhì)譜儀建立46 種皂苷類成分的檢測方法,并對人參不同加工品中皂苷類化合物的含量和組成進行檢測分析和比較。該方法進一步拓展了現(xiàn)有方法的檢測范圍,增加了樣品前處理的凈化技術(shù)和皂苷類化合物的檢測種類,操作簡便、快速,為不同人參加工品中多種皂苷類化合物的快速分析檢測提供了技術(shù)參考。
1 材料與方法
1.1 材料與試劑
3 種人參加工品(生曬參、紅參和黑參)為吉林省撫松萬良人參市場隨機購買。
46 種皂苷標準物質(zhì)對照品(純度均不小于95%)購于上海源葉生物科技有限公司和成都曼斯特生物科技有限公司。
甲醇、乙腈(均為色譜純) 德國Merck公司;甲酸(色譜純) 美國Sigma-Aldrich公司;其他試劑均為分析純。
N-丙基乙二胺(primary secondary amine,PSA)、十八烷基鍵合硅膠(C18)吸附劑、石墨化碳黑(graphitized carbon black,GCB)吸附劑 北京恩加壹科技有限公司;實驗用水為超純水(Milli-Q純水處理系統(tǒng)制備,美國Millipore公司)。
1.2 儀器與設備
Sep-pak C18固相萃取小柱(1 g/6 mL) 美國Waters公司;LC-30A超高效液相色譜儀 日本Shimadzu公司;AB QTRAP 4500三重四極桿/復合線性離子阱質(zhì)譜儀、Analyst、Peak View和MultiQuant數(shù)據(jù)處理系統(tǒng) 美國AB SCIEX公司;Kinetex XB-C18色譜柱(2.1 mm ×100 mm,2.6 μm) 美國Phenomenex公司;KQ-500DE數(shù)控超聲波清洗器 昆山市超聲儀器有限公司;Heraeus Multifuge XIR臺式冷凍離心機 美國Thermo公司;AL204型電子天平 美國Sartorius公司;VORTEX-6旋渦混合器 北京海天友誠科技有限公司。
1.3 方法
1.3.1 標準溶液的配制
準確稱取4 6 種皂苷標準品各1 0 m g(精確至0.1 mg),分別以甲醇溶解并定容,配制成1 mg/mL的標準品儲備液,于4 ℃避光保存?zhèn)溆谩H∵m量標準儲備液,以甲醇稀釋成100 μg/mL的標準中間液。分別取適量標準中間液,根據(jù)各化合物的響應情況,分別用甲醇稀釋成不同質(zhì)量濃度的系列混合標準溶液。
1.3.2 樣品前處理
提取:取干燥至恒質(zhì)量的人參加工品樣品粉碎,過60 目篩,準確稱取1 g(精確到0.000 1 g)樣品粉末,加入50 mL 80%甲醇溶液,浸泡過夜,超聲40 min,以5 000 r/min離心15 min,精密移取上清液2 mL,60 ℃下氮氣吹干,用甲醇溶解并定容至5 mL,待凈化。
凈化:準確移取提取液2 mL至裝有100 mg PSA的離心管中,渦旋振蕩2 min,5 000 r/min離心6 min。上清液過0.22 μm濾膜,備用。或根據(jù)實際濃度用甲醇適當稀釋至線性范圍內(nèi),作為供試品溶液待測。
1.3.3 色譜條件
Kinetex XB-C18色譜柱(2.1 mm×100 mm,2.6 μm);柱溫40 ℃;流速0.3 mL/min;進樣量2.0 μL;流動相A為0.1%甲酸溶液,B為乙腈;梯度洗脫:0~1.0 min,81% A、19% B;1.0~2.0 min,81%~72% A、19%~28% B;2.0~14.0 min,72%~65% A、28%~35% B;14.0~20.0 min,65%~55% A、35%~45% B;20.0~25.0 min,55%~43% A、45%~57% B;25.0~28.0 min,43%~0% A、57%~100% B;28.0~30.0 min,0% A、100% B;30.0~32.0 min,0%~81% A、100%~19% B;32.0~35.0 min,81% A、19% B。
1.3.4 質(zhì)譜條件
電噴霧離子源負離子模式;MRM模式;霧化電壓-4 500 V;離子化溫度450 ℃;霧化氣壓力45 psi;輔助氣壓力4 5 p s i;碰撞氣壓力M e d i u m;氣簾氣壓力35 psi。
1.3.5 基質(zhì)效應計算
基質(zhì)效應(matrix effect,ME)/%=A/B×100。其中A為樣品中皂苷類物質(zhì)的峰面積;B為純?nèi)軇┲型瑵舛仍碥疹愇镔|(zhì)對應的峰面積。當ME>1時,表示基質(zhì)增強效應,當ME<1時,表示基質(zhì)抑制效應。若ME為80%~120%時,表示ME不明顯,可以忽略;若ME在50%~80%或120%~150%時,表現(xiàn)為中等ME;若ME<50%或者ME>150%時,則表現(xiàn)為強ME[25]。
1.4 數(shù)據(jù)處理
通過與儀器配套的AB SCIEX公司的Analyst 1.6軟件系統(tǒng)完成數(shù)據(jù)采集與處理,在Multi Quant 3.0和Peak View 2.0軟件上定性定量處理和譜圖分析,采用Excel和分析軟件SIMCA 14.1對結(jié)果進行統(tǒng)計分析及圖表繪制。
2 結(jié)果與分析
2.1 色譜條件的優(yōu)化
本實驗所測的皂苷類化合物種類多,但結(jié)構(gòu)相似,性質(zhì)差異較小,且含有多對同分異構(gòu)體,這些化合物具有相同的前體和產(chǎn)物離子,在質(zhì)譜上無法通過離子對其進行區(qū)分,若要準確定量這些化合物,色譜峰要有一定程度的分離。因此實驗對色譜條件進行優(yōu)化,以實現(xiàn)皂苷類化合物的最佳色譜分離以及各化合物的良好峰形和最佳響應。實驗首先考察了在相同參數(shù)條件下,不同型號色譜柱Waters HSS T3(100 mm×2.1 mm,1.7 μm)、Waters BEH C18(100 mm×2.1 mm,1.7 μm)和Kinetex XB-C18(2.1 mm×100 mm,2.6 μm)的分離效果,結(jié)果表明,HSS T3色譜柱對于部分皂苷類物質(zhì)分離效果較差,特別是無法實現(xiàn)同時對多對同分異構(gòu)體化合物的基線分離;而Waters BEH C18和Kinetex XB-C18色譜柱對46 種目標化合物均呈現(xiàn)較好的色譜峰形和分離度,但考慮到對系統(tǒng)壓力的影響,選擇具有低柱壓的Kinetex XB-C18作為分離色譜柱。在流動相選擇方面,考察反相色譜常用的有機相溶劑(甲醇、乙腈)與水組成的流動相體系對皂苷類成分的分離效果,發(fā)現(xiàn)以乙腈體系為流動相時,系統(tǒng)平衡時間相對較快,且洗脫能力和分離效果均優(yōu)于甲醇體系。在流動相中加入適量的甲酸可以減少色譜峰的拖尾,提高皂苷類化合物的離子化效率,改善色譜峰形,并增強目標化合物的響應信號和穩(wěn)定性。因此,本實驗流動相選擇最常用的乙腈-0.1%甲酸溶液系統(tǒng),在很大程度上減少了溶劑系統(tǒng)帶來的背景和基質(zhì)干擾。最后,通過調(diào)節(jié)梯度洗脫程序使互為同分異構(gòu)體的皂苷類化合物進一步分離,由待測目標化合物的結(jié)構(gòu)可知,本研究中46 種皂苷類化合物中同分異構(gòu)體較多,但可通過保留時間和離子碎片的不同加以區(qū)分。例如,人參皂苷Rb2、Rb3和Rc與人參皂苷Rs1和Rs2這兩組同分異構(gòu)體,它們各自產(chǎn)生的檢測離子碎片雖然相同,但可通過保留時間的不同得到較好分離;而另一組同分異構(gòu)體原人參三醇型皂苷Rf和擬人參皂苷F11,二者保留時間較接近,通過調(diào)節(jié)流動相梯度也無法得到較好分離,但由于二者產(chǎn)生的離子碎片不同則可加以區(qū)分。在優(yōu)化的儀器條件下,各皂苷類化合物靈敏度和和峰形均可滿足檢測要求,整個梯度運行時間為35 min,46 種皂苷類化合物均得到有效分離,能夠達到準確定性定量目的。46 種目標物混合標準溶液的MRM色譜圖見圖1,46 種化合物信息見表1。
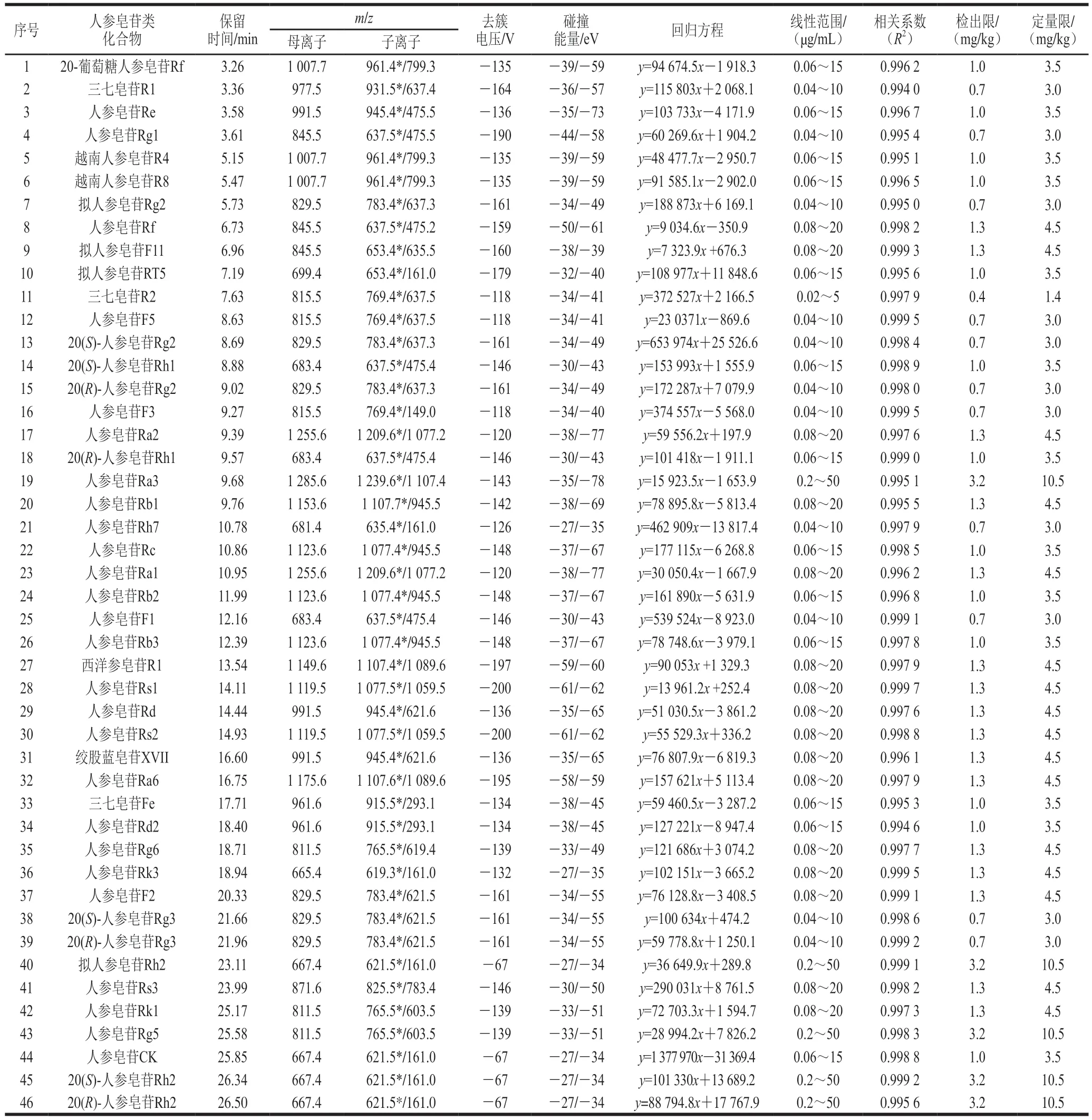
表1 46 種人參皂苷類化合物的質(zhì)譜參數(shù)、線性關(guān)系、檢出限和定量限Table 1 Mass spectrometric parameters, linearities, LODs and LOQs of 46 ginsenoside compounds
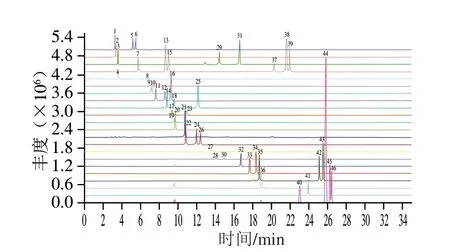
圖1 46 種人參皂苷類化合物標準物質(zhì)的MRM色譜圖Fig.1 MRM chromatograms of 46 ginsenoside standards
2.2 質(zhì)譜條件的優(yōu)化
采用針泵直接將標準溶液注入離子源的方式對7 種皂苷類化合物質(zhì)譜參數(shù)進行優(yōu)化。分別將46 種待測皂苷類化合物標準溶液(質(zhì)量濃度均為1.0 μg/mL)以7 μL/min注入電噴霧離子源,采用正、負2 種離子模式進行掃描,發(fā)現(xiàn)皂苷類化合物在負離子模式下具有較高的穩(wěn)定性和較高的響應值,這與文獻[26]相關(guān)報道一致,因此選擇負離子模式。皂苷類物質(zhì)在負離子模式下的一級質(zhì)譜圖中,其準分子離子主要以[M-H]-和[M+HCOOH-H]-的形式存在。在Q1進行全掃描中,找出母離子,母離子在Q2打碎后,在Q3進行子離子全掃描以確定各組分的主要碎片離子,選擇2 個無干擾、靈敏度相對較高的穩(wěn)定的特征子離子,組成MRM離子對,作為定性定量的離子對。在MRM模式下優(yōu)化去簇電壓和碰撞能量,最終確定了46 種皂苷類化合物的MRM離子對、去簇電壓及碰撞能量。優(yōu)化后的質(zhì)譜參數(shù)如表1所示。
2.3 提取條件的選擇
人參皂苷類化合物的提取相關(guān)報道較多,常用的提取溶劑和方法有甲醇回流提取[27]、甲醇超聲提取[28]、80%甲醇超聲提取[12]、水飽和正丁醇超聲提取[29]和60%乙醇超聲提取[14]。本研究在前人研究的基礎(chǔ)上,選用提取方法較簡單的超聲提取法,并考察甲醇、80%甲醇、水飽和正丁醇和60%乙醇作為提取溶劑時,各個皂苷類化合物的提取效果。經(jīng)過對比發(fā)現(xiàn),80%甲醇和60%乙醇對大部分皂苷類化合物的提取效率優(yōu)于純甲醇和水飽和正丁醇,但60%乙醇為提取溶劑時,提取液的顏色較濃,雜質(zhì)較多,且基線不穩(wěn)定,不利于后續(xù)的凈化處理,而80%甲醇溶液的提取效果總體最佳,基線和峰形也較好,因此最終選用80%甲醇溶液作為提取溶劑。
2.4 凈化條件的優(yōu)化
人參加工品提取液中的糖類、色素及有機酸等雜質(zhì)會產(chǎn)生ME,干擾待測物的分析,而且會損壞檢測儀器和色譜柱,需要對樣品進一步凈化。為了使凈化時盡可能多地去除雜質(zhì)干擾,又能減少對目標物的損失,同時使實驗方法達到簡單、快速、準確,具有時效性。實驗對Sep-pak C18(1 g/6 mL)為固相萃取柱的固相萃取法[9]和分散固相萃取2 種凈化方式進行比較。發(fā)現(xiàn)C18固相萃取柱凈化后,各化合物的回收率可達到80%~105%,凈化效果也較明顯,但C18固相萃取柱凈化過程需要活化、平衡、洗脫等多個步驟,處理時間長,過程也較繁瑣。分散固相萃取凈化方法具有快速、簡單、廉價、有效等特點,可將提取液直接達到凈化目的,是較為理想的樣品前處理凈化方法。常用的吸附劑有C18、GCB、PSA等。實驗分別考察3 種吸附劑C18、GCB和PSA對46 種皂苷類化合物回收率的影響,結(jié)果如圖2所示,當使用C18作為吸附劑時,46 種目標化合物的回收率為8.9%~31.6%,這是因為C18吸附劑具有疏水作用,除了能吸附色素、脂肪和維生素等非極性的組分,對中等極性或極性小的分子也有吸附[30],因此對皂苷類化合物的吸附作用較強,不利于凈化。當使用GCB作為吸附劑時,凈化后的提取液顏色最淺,雖然對人參樣品提取液中色素類物質(zhì)凈化效果最好,但由于GCB其表面光滑無孔的石墨晶型結(jié)構(gòu)決定其對非極性化合物、弱極性化合物和部分極性化合物都具有一定的吸附作用[31]。因此GCB不僅對色素類物質(zhì)吸附性好,還會吸附目標化合物,使目標化合物的回收率降低,GCB對46 種目標化合物凈化后的回收率為30.1%~69.5%。PSA對46 種目標化合物的吸附作用均較弱,回收率在80.2%~103.8%之間,而且PSA對脂肪酸、色素、有機酸、及糖等極性干擾物的去除效果較好,基本可滿足凈化需求[32]。
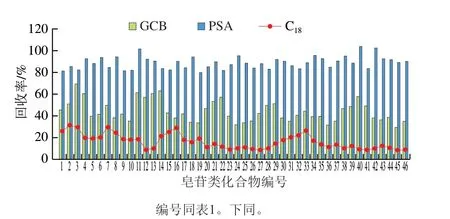
圖2 3 種吸附劑對46 種目標化合物回收率的影響Fig.2 Effects of three sorbents on recoveries of 46 analytes
為了進一步提高樣品的凈化效果,實驗對PSA用量進行調(diào)整,在2 mL提取液中分別加入60 、80、100、120、140 mg的吸附劑PSA進行凈化處理,同樣以凈化回收率作為考察指標進行比較,結(jié)果如圖3所示,發(fā)現(xiàn)PSA添加量為60、80、100 mg時46 種皂苷類化合物的回收率無明顯變化,回收率為81.9%~113.5%,其中100 mg PSA的回收率和凈化效果均較好。隨著PSA用量的增加,目標化合物的回收率受到嚴重影響,添加量120 mg PSA的回收率為72.2%~85.8%,添加量140 mg PSA的回收率為65.6%~80.8%,這是由于吸附劑用量過高時,會對目標化合物產(chǎn)生部分吸附,從而降低目標化合物的回收率。因此,綜合考慮回收率和凈化效果,實驗最終選擇2 mL提取液中加入100 mg PSA作為凈化劑,可以獲得良好的凈化效果,基質(zhì)干擾小,且不會對待測皂苷類化合物產(chǎn)生明顯吸附。
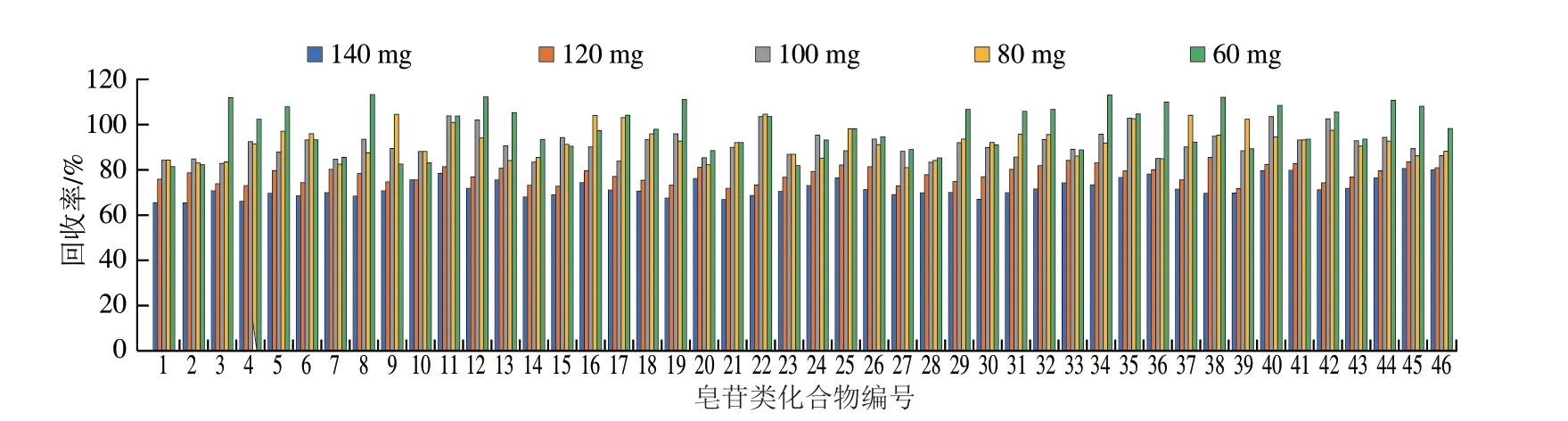
圖3 PSA吸附劑用量對46 種目標化合物回收率的影響Fig.3 Effect of PSA dosage on recoveries of 46 analytes
2.5 ME考察
由于人參加工樣品基質(zhì)復雜,可能會對檢測結(jié)果的準確度造成干擾,為此對方法中的ME進行考察。分別以生曬參、紅參、黑參樣品為基質(zhì),按照1.3.2節(jié)樣品前處理方法制備基質(zhì)溶液,分別用基質(zhì)溶液和純?nèi)軇┡渲埔幌盗谢旌匣|(zhì)標準工作溶液和純?nèi)軇藴使ぷ饕海糜跇藴是€的測定。利用基質(zhì)標準曲線測算出基質(zhì)中目標物質(zhì)本底濃度和峰面積,再用純?nèi)軇藴是€測算出對應濃度下純?nèi)軇┲心繕宋镔|(zhì)的峰面積,利用公式計算ME。
本研究中分別比較了樣品在分散固相萃取法凈化前后的ME,如圖4所示,分散固相萃取凈化前生曬參、紅參、黑參樣品中部分目標化合物的ME值在120%~150%之間,表現(xiàn)為中等基質(zhì)增強效應;部分目標化合物的ME值在50%~80%之間,表現(xiàn)為中等基質(zhì)抑制效應。經(jīng)分散固相萃取凈化后,46 種目標化合物在生曬參、紅參、黑參樣品中的ME值分別為88.6%~110.9%、86.5%~109.6%和84.8%~112.2%,ME表現(xiàn)不明顯,說明樣品基質(zhì)經(jīng)過前處理過程的分散固相萃取凈化和較大體積的稀釋,有效降低了人參加工品中ME的影響,保證了定性定量結(jié)果的準確、可靠。
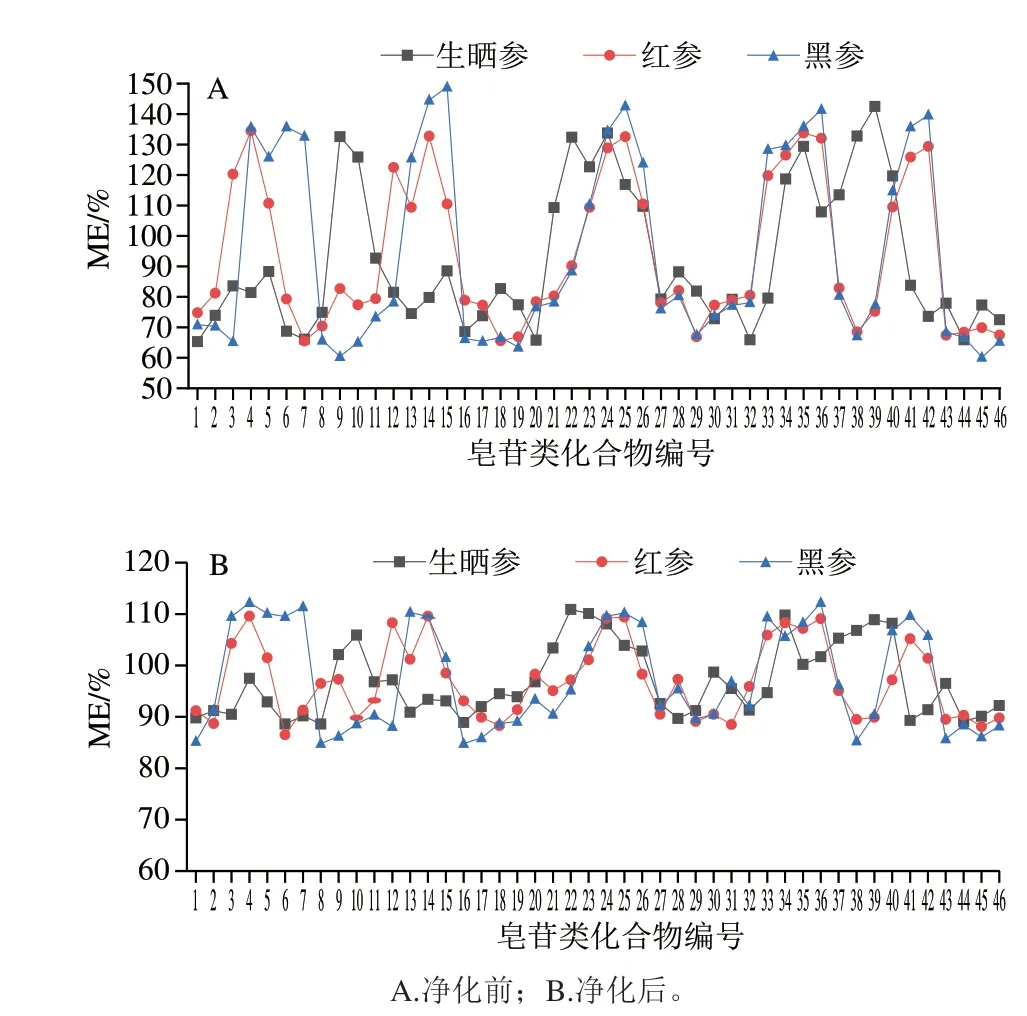
圖4 46 種目標化合物MEFig.4 Matrix effects of 46 analytes
2.6 方法學驗證
2.6.1 方法的線性范圍、檢出限及定量限結(jié)果
在優(yōu)化的分析條件下,測定46 種皂苷類化合物系列混合標準工作液,以各目標化合物定量離子對的峰面積為縱坐標(y),以相應的質(zhì)量濃度為橫坐標(x),繪制標準曲線,得到各目標物的線性回歸方程。以3 倍信噪比確定方法的檢出限,以10 倍信噪比確定方法的定量限。46 種人參皂苷類化合物在0.02~50 μg/mL范圍內(nèi)呈現(xiàn)較好的線性關(guān)系,所對應的相關(guān)系數(shù)(R2)均大于0.99。方法的檢出限和定量限分別在0.4~3.2 mg/kg和1.4~10.5 mg/kg范圍內(nèi),均滿足檢測需要,結(jié)果見表1。
2.6.2 回收率實驗結(jié)果
在已知皂苷類化合物含量的生曬參、紅參和黑參樣品中,分別添加低、中、高3 個濃度水平的加標回收實驗,每個添加水平重復5 次。計算平均回收率和相對標準偏差(relative standard deviation,RSD),結(jié)果見表2。46 種皂苷類化合物除20(S)-人參皂苷Rh2和20(R)-人參皂苷Rh2在3 種人參加工產(chǎn)品中平均加標回收率為80.5%~84.6%,其余44 種皂苷類化合物的平均加標回收率均達到85.2%~111.6%之間,RSD均在1.3%~6.8%之間,表明本實驗方法有較好的回收率,結(jié)果準確可靠。
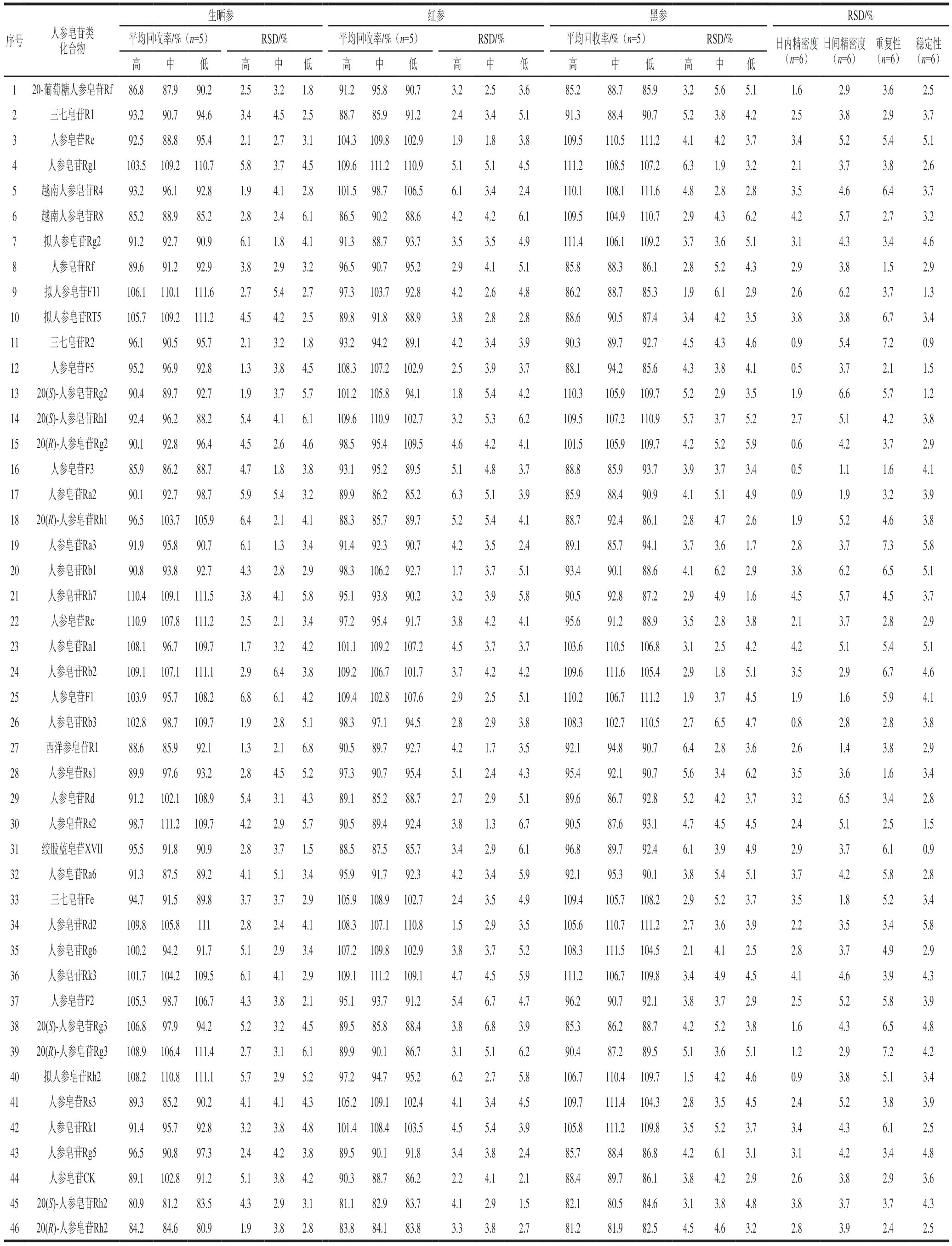
表2 皂苷類化合物的加標回收率、日內(nèi)和日間精密度、重復性及穩(wěn)定性Table 2 Spiked recoveries, intra-day and intra-day precision, repeatability and stability for the determination of 46 ginsenosides
2.6.3 精密度、穩(wěn)定性和重復性結(jié)果
取5 μg/mL的混合標準溶液分別在1 d內(nèi)(6 次)和3 d內(nèi)重復進樣(2 次/d)。計算各成分含量的日內(nèi)和日間RSD,用以表征日內(nèi)和日間精密度。取同一樣品分析溶液,分別在0、2、4、8、12 h和24 h進樣6 次,測定目標化合物峰面積的RSD,考察其穩(wěn)定性。取同一批樣品粉末6 份,按1.3.2節(jié)方法制備供試品溶液,在1.3.3節(jié)和1.3.4節(jié)條件下進樣,計算目標化合物測得含量的RSD,考察其重復性。結(jié)果見表2。46 種皂苷類化合物的日內(nèi)精密度和日間精密度分別為0.5%~4.5%、1.1%~6.6%。重復性RSD范圍為1.5%~7.3%,穩(wěn)定性RSD范圍為0.9%~5.8%,表明該測定方法具有較高的精密度和較好的穩(wěn)定性和重復性。
2.7 實際樣品分析
應用本方法對市售的15 批生曬參、紅參和黑參3 種不同人參加工品進行分析檢測,每種人參加工品5 批,每個樣品重復測定3 次(圖5)。皂苷類成分在3 類人參加工品中呈現(xiàn)明顯不同的含量分布特征,結(jié)果如表3所示。為了更直觀地區(qū)分不同加工方式的人參產(chǎn)品,以46 種人參皂苷的含量為變量,采用多元變量統(tǒng)計分析軟件SIMCA 14.1對15 份不同人參加工品進行主成分分析(principle component analysis,PCA),見圖6。PC1和PC2的累計貢獻率達到95.4%,可代表皂苷類成分指標。得分圖中生曬參、紅參和黑參樣品之間相距較遠,彼此之間可獲得良好區(qū)分,表明不同加工方式的人參產(chǎn)品皂苷類成分的含量差異較大。
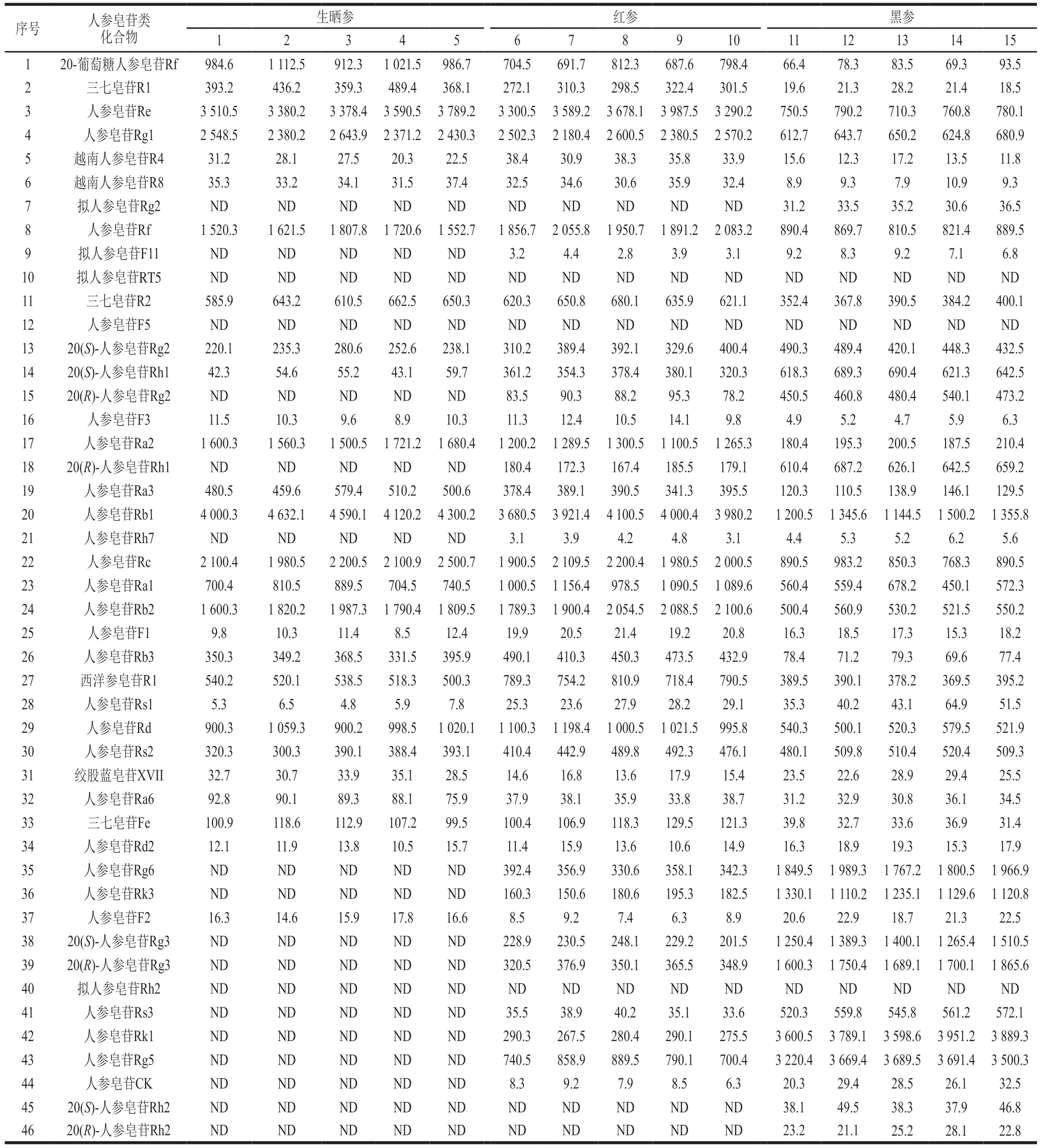
表3 3 種人參加工產(chǎn)品中人參皂苷類化合物的含量(n=3)Table 3 Contents of ginsenosides in three processed ginseng products (n = 3)mg/kg

圖5 代表性樣品的總離子流色譜圖Fig.5 Total ion current chromatograms of representative samples
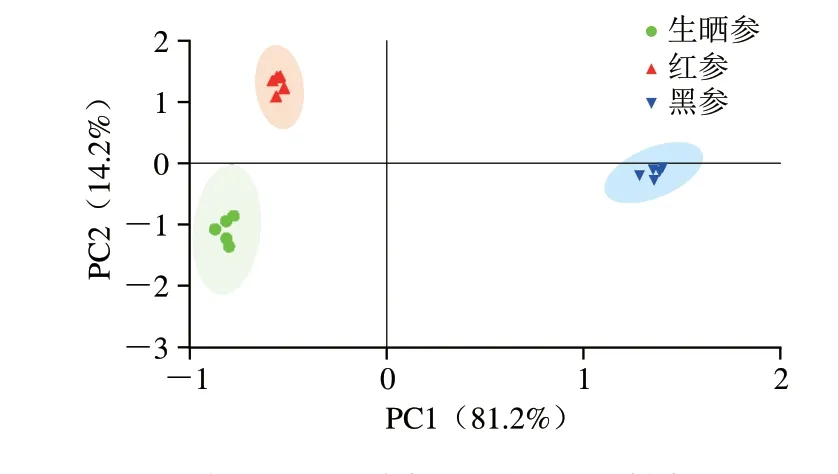
圖6 3 種人參加工產(chǎn)品的PCA得分Fig.6 PCA score plot for three processed ginseng products
生曬參是通過在陽光下使鮮人參脫水生產(chǎn),而紅參和黑參是通過不同的高溫循環(huán)蒸制生產(chǎn)[33-34],這些不同的加工方法誘導了人參皂苷的化學變化,部分人參皂苷轉(zhuǎn)化成了稀有人參皂苷[35]。因此,加工方式的不同是導致人參產(chǎn)品中皂苷類成分含量差異較大的主要原因。
為了進一步明確不同加工方式的人參差異性成分,在PCA基礎(chǔ)上,采用變量投影重要性(variable importance in projection,VIP)法篩選,VIP值見圖7。以VIP值大于1為標準[36],篩選出造成不同加工方式人參差異的17 個皂苷成分,包括20-葡萄糖人參皂苷Rf、西洋參皂苷R1、人參皂苷Re、Rg1、Rf、Rh1(S)、Ra2、Rb1、Rc、Ra1、Rb2、Rg6、Rk3、Rg3(S)、Rg3(R)、Rk1、Rg5。如圖8所示,20-葡萄糖人參皂苷Rf、人參皂苷Re、Rg1、Rf、Ra2、Rb1、Rc和Rb2在生曬參和紅參中相對含量較高,在黑參中相對含量較低,這些皂苷均為常量人參皂苷,可作為生曬參和紅參的主要成分;而人參皂苷Rh1(S)、Rg6、Rk3、Rg3(S)、Rg3(R)、Rk1、Rg5在黑參中相對含量較高,在紅參中相對含量較低,在生曬參中幾乎不含,且這些皂苷均為稀有人參皂苷,可見經(jīng)過蒸制的紅參和黑參樣品中部分原有的人參皂苷發(fā)生了轉(zhuǎn)化,產(chǎn)生了稀有人參皂苷,這些差異性皂苷成分可作為區(qū)分黑參、紅參和生曬參的標志物群。
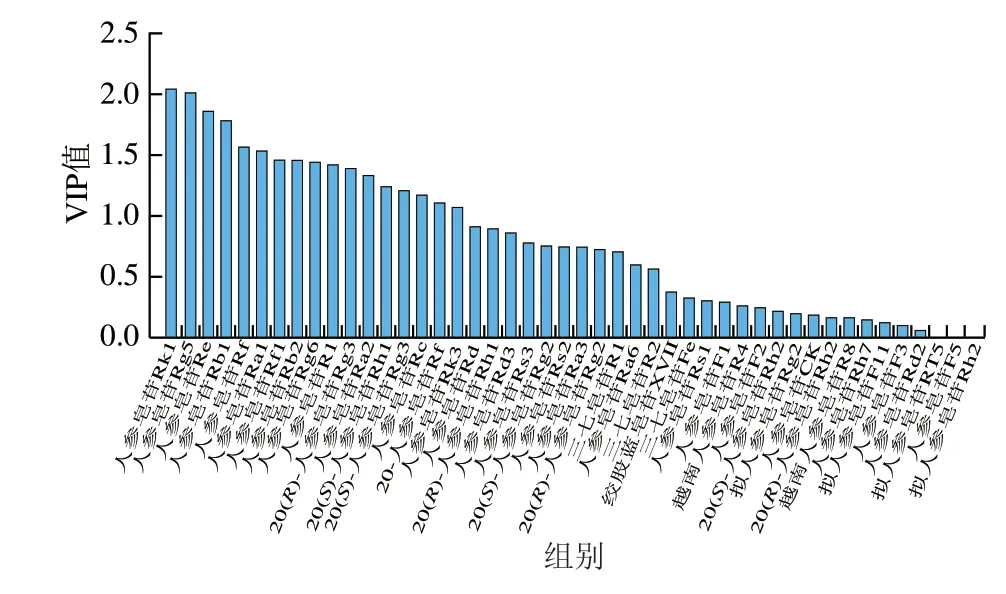
圖7 各成分的正交偏最小二乘判別分析VIP值圖Fig.7 Orthogonal partial least squares-discriminant analysis VIP plot for 46 ginsenosides
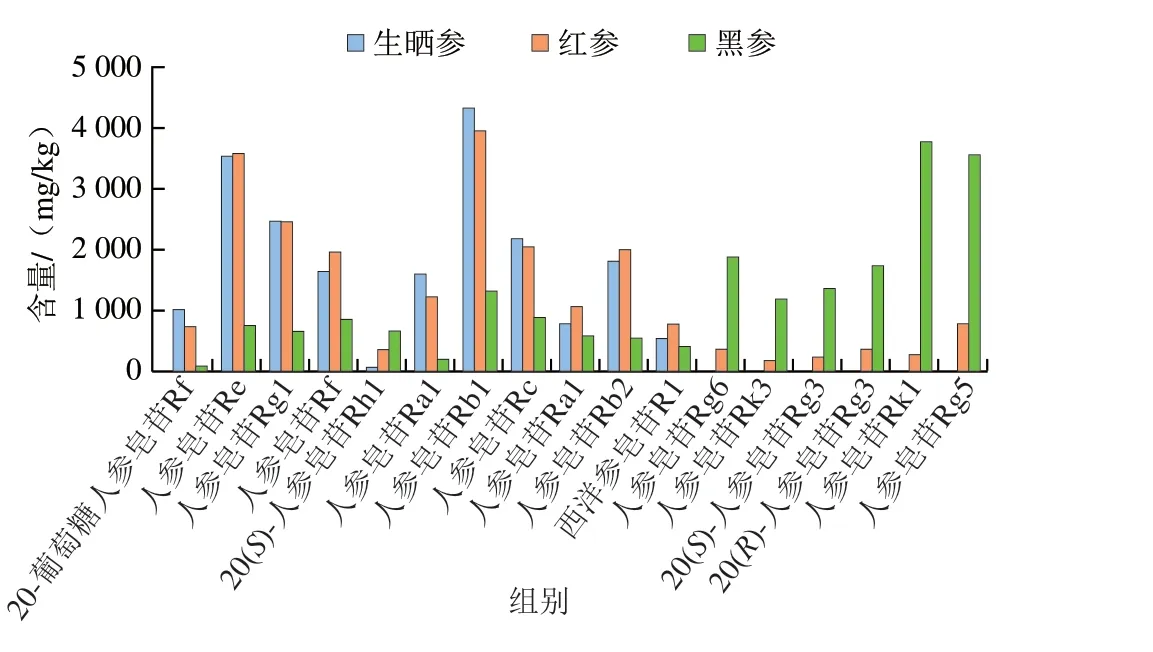
圖8 3 種人參加工品中差異性成分含量Fig.8 Contents of differential ginsenosides in three processed ginseng products
3 結(jié) 論
本研究采用分散固相萃取凈化,建立了人參加工品中46 種皂苷類化合物的UPLC-MS/MS檢測方法。實現(xiàn)在35 min內(nèi)可同時完成46 種皂苷類化合物的快速分析和定量檢測,與已有方法相比,本方法引入了分散固相萃取技術(shù),即降低了基質(zhì)干擾,又能減少對目標物的損失,且簡化了操作步驟,使樣品前處理更加簡便高效,實用性強。通過儀器條件的優(yōu)化,采用MRM檢測,在很大程度上提高了皂苷類化合物的檢測通量,可有效縮短分析檢測時間,提高工作效率。通過方法學驗證,證明該法具有準確快速、重復性好且雜質(zhì)干擾小等特點。經(jīng)3 類人參加工品生曬參、紅參和黑參共15 批實際樣品的檢測分析驗證,完全能夠滿足檢測需求,可應用于實際人參加工品中皂苷類化合物的直接測定。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數(shù)理化·七年級數(shù)學人教版(2021年6期)2021-11-22 07:50:58
中學生數(shù)理化·七年級數(shù)學人教版(2021年6期)2021-11-22 07:50:58
中學生數(shù)理化·七年級數(shù)學人教版(2021年6期)2021-11-22 07:50:58
中學生數(shù)理化·七年級數(shù)學人教版(2020年12期)2021-01-18 06:57:46
中學生數(shù)理化·七年級數(shù)學人教版(2020年12期)2021-01-18 06:57:46
中學生數(shù)理化·七年級數(shù)學人教版(2019年9期)2019-11-25 07:34:36
中學生數(shù)理化·七年級數(shù)學人教版(2019年9期)2019-11-25 07:34:34
中學生數(shù)理化·七年級數(shù)學人教版(2019年12期)2019-05-21 02:53:50
中學生數(shù)理化·七年級數(shù)學人教版(2019年12期)2019-05-21 02:53:48
