KCNQ2基因相關癲癇臨床表型與基因型相關性研究
2023-11-08 11:09:34陳亞楠何云燕趙婷周瑤王娜馮燕燕韓雄
河南醫學研究 2023年20期
陳亞楠,何云燕,趙婷,周瑤,王娜,馮燕燕,韓雄
(1.河南省人民醫院神經內科,鄭州大學人民醫院,河南 鄭州 450003;2.青島大學附屬婦女兒童醫院 神經內科,山東 青島 266000)
KCNQ2基因位于染色體20q13.3,包含17個外顯子,編碼電壓門控鉀通道,在胎兒和成人大腦中均有表達[1],是最常見的引起癲癇的基因之一。Singh等[2]于1998年首次報道了KCNQ2基因是良性家族性新生兒癲癇(benign familial neonatal epilepsy,BFNE)的致病基因。2003年Dedek等[3]首先報道了早發型癲癇性腦病(early onset epileptic encephalopathy,EOEE)患者中KCNQ2的致病突變。隨著基因檢測技術的發展,越來越多關于KCNQ2與癲癇相關的研究被報道。KCNQ2基因的突變已證明與多種類型癲癇相關,如肌陣攣發作、局灶性發作、全面強直-陣攣發作、BFNE、良性家族性新生兒-嬰兒癲癇、良性家族性嬰兒癲癇(benign familial infantile epilepsy,BFIE)、早發癲癇性腦病(early infantile epileptic encephalopathy,EIEE)、嬰兒痙攣癥(west syndrome,WS)、大田原綜合征(ohtahara syndrome,OS)等[4-7]。目前報道的關于KCNQ2相關的突變已超過上百個,雖有文獻報道其基因型與癲癇發作的相關性,但缺乏比較全面的總結分析。本研究進一步系統總結了已知KCNQ2的突變及癲癇相關疾病,旨在闡明突變類型、突變位置(分子亞區)與臨床表型之間的關系。
1 對象與方法
1.1 研究對象
從人類基因突變數據庫(Human Gene Mutation Database,HGMD)專業版(http://www.hgmd.cf.ac.uk/ac/index.php)、PubMed數據庫(http://www.ncbi.nlm.nih.gov/pubmed/)、中國知網及萬方數據庫系統檢索截至2022年9月的所有KCNQ2突變(451個)和對應疾病及相關表型。同時根據UniProt數據庫收錄的人源KCNQ2蛋白對其結構域進行劃分。
1.2 方法
為了探討突變類型和臨床表型之間的關系,本研究將KCNQ2突變分為兩大類,破壞性突變(無效突變)和錯義突變。無效突變是那些導致蛋白嚴重畸形并導致功能喪失和單倍功能不足的突變[8-9],包括截短突變(無義和框移)、經典剪接位點突變(標準±1或2)、起始密碼子突變或單/多外顯子缺失。癲癇表型的譜系潛在范圍從輕度癲癇如局灶性癲癇到嚴重的發育性癲癇性腦病。將癲癇的表型分為輕表型與重表型。輕表型包括肌陣攣癲癇、全面性癲癇、良性嬰兒性癲癇、良性新生兒癲癇、良性新生兒-嬰兒癲癇和早發型癲癇;重表型包括癲癇合并神經系統發育障礙、癲癇合并孤獨癥、發育性癲癇性腦病、癲癇性腦病、早發性嬰兒性癲癇性腦病、新生兒癲癇性腦病、癲癇性腦病7型、WS和OS。為了便于分析分子亞區域與癲癇表型的關系,將KCNQ2的分子亞區分為N/C末端、S1~S3區、S4電壓感應區(voltage-sensing domain,VSD)、D-Link區、孔區(S5~S6)。根據數據庫及檢索到的KCNQ2相關的突變位點進行亞區分類,并對癲癇表型進行分類,從而進行統計分析。
1.3 統計學分析
使用SPSS 26.0軟件進行統計分析。采用χ2比較不同突變類型中癲癇輕重表型之間的差異、各分子亞區內癲癇輕表型與重表型之間的差異。P<0.05為差異有統計學意義。
2 結果
2.1 KCNQ2基因突變導致的疾病
本研究檢索到的451個突變所致疾病中,非神經系統疾病3例(肌張力障礙1例,擴張型心肌病1例,先天性心臟病1例),非癲癇的神經系統相關疾病12例(周圍神經高興奮性1例,神經系統異常2例,神經系統發育障礙9例)。癲癇相關的突變436例(96.67%,其中癲癇輕表型223例,重表型213例)。
2.2 KCNQ2基因突變類型與癲癇臨床表型的相關性
KCNQ2基因突變類型中癲癇相關的錯義突變255個(58.49%),框內突變20個(4.59%),無義突變26個(5.96%),拷貝數突變(copy number variation,CNV)51個(11.70%),移碼突變59個(13.53%),剪切位點突變25個(5.73%,其中經典剪切位點突變20個,非經典剪切位點突變5個)(見表1)。錯義突變中癲癇輕表型有94個,重表型有161個,在錯義突變中更常見表型為癲癇重表型(P<0.05)(見圖1A)。在破壞性突變(無效突變)中癲癇輕表型112個,重表型44個,在無效突變中更容易出現輕表型,且出現輕表型的概率與重表型之間的差異有統計學意義(P<0.05)(見圖1B)。
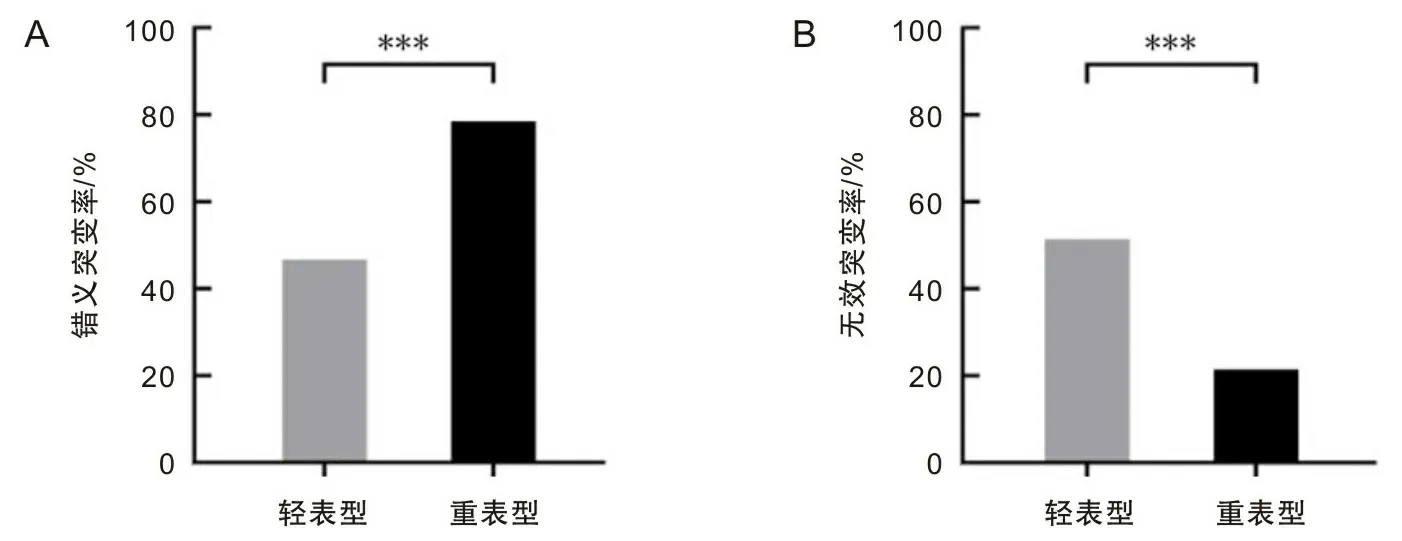
A為KCNQ2基因錯義突變與癲癇表型的相關性;B為KCNQ2無效突變與癲癇表型的相關性;***P<0.001。
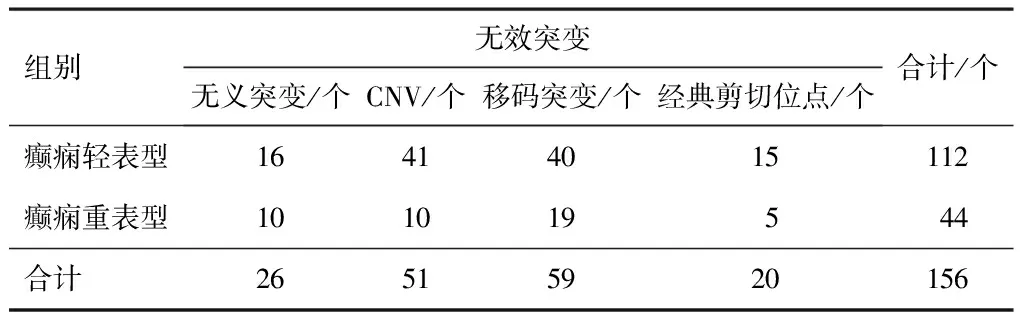
表1 無效突變中各種突變類型的數量
2.3 KCNQ2基因分子亞區與癲癇表型的關系
在錯義突變方面,輕表型患者中50%(47/94)位于N/C末端,重表型患者中31.05%(50/161)位于N/C末端,差異有統計學意義(P<0.05)。輕表型患者中25.53%(24/94)位于S5~S6區,重表型患者中43.48%(70/161)位于S5~S6區,差異有統計學意義(P<0.05)。其余3個區分布情況相比,差異無統計學意義(P>0.05)。見表2。
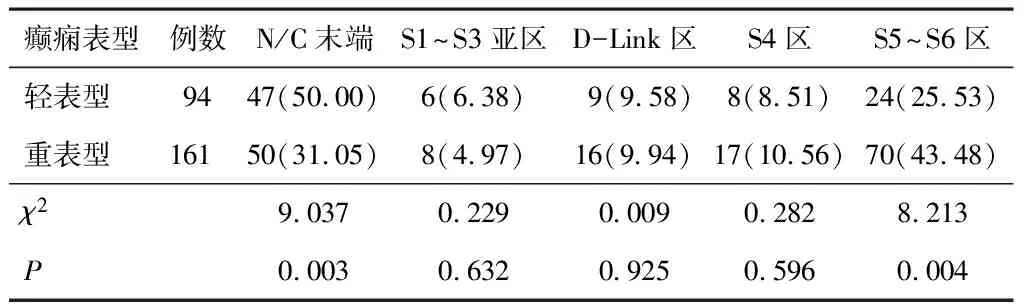
表2 KCNQ2基因分子亞區與癲癇表型的關系[n(%)]
3 討論
在目前已知的遺傳性癲癇中,同一基因的致病突變可能導致不同甚至相反的癲癇表型,例如,KCNQ2基因可以導致自限性癲癇(也稱為良性癲癇表型)[10]或發育性癲癇(嚴重癲癇表型)[11-12]。目前出現這種現象的機制尚不明確。臨床上最常見的例子就是BFNE和KCNQ2相關的發育性癲癇性腦病,這兩者均由KCNQ2基因引起[13]。
KCNQ2相關疾病代表了由KCNQ2雜合致病突變引起的一系列疾病。KCNQ2相關疾病的臨床特征包括從輕度KCNQ2相關的BFNE,到重度KCNQ2相關的新生兒癲癇性腦病[13]。其中也包含少數的非神經系統疾病及非癲癇的神經系統疾病,本文總結了目前所報道的關于KCNQ2的451個突變,在所致疾病中,癲癇相關的突變占96.67%,非神經系統疾病占0.66%,非癲癇的神經系統相關疾病占2.67%。
導致KCNQ2單倍體功能不全的突變與自限性家族性新生兒癲癇相關[14],而更嚴重的通道功能抑制和/或軸突亞細胞定位的預防與導致新生兒發病和癲癇性腦病表型的致病突變有關[15]。本研究中將不同突變類型中出現癲癇輕表型與重表型的概率進行總結分析,其中錯義突變最常見,與之前文獻報道的突變類型[16-18]一致。一般來說,由單個KCNQ2等位基因(無義、剪接或移碼突變)功能喪失引起的單倍劑量不足是家族性KCNQ2-BFNE最常見的原因,單倍體劑量不足常導致電壓門控鉀通道出現輕度功能喪失,從而更容易表現為癲癇輕表型[19]。迄今為止,KCNQ2-NEE中發現的致病性突變均為新生錯義變體,而錯義突變被認為可能導致電壓門控鉀通道出現更嚴重的功能缺陷從而更容易出現癲癇重表型[20]。本研究也發現錯義突變導致的癲癇重表型概率更高,在移碼突變與整個基因的缺失或重復所導致的突變中癲癇輕表型出現的概率較高。
兩個KCNQ2和兩個KCNQ3亞單元組裝成1個M型鉀通道,產生M電流,M電流是一種超極化電流,其活性通過除極增強,從而防止多種動作電位的失控[21]。KCNQ2的結構由1個短的胞質N端(91個氨基酸)、6個跨膜段(S1~S6)和1個P環,以及1個長C端(559個氨基酸)與磷脂酰肌醇二磷酸酯和鈣調蛋白結合域組成。S4段包含通道的VSD,S5和S6段以及它們之間的P環(也稱H5)形成了離子通道的孔區,它們的構象變化調節了異四聚體選擇性的開放[18],目前認為胞內C端參與了Kv2的細胞內定位、鉀通道組裝和膜磷脂調控,與鈣調蛋白等蛋白之間相互作用[22]。
本研究中錯義突變位點在S1~S3、D-Link區、S4區域中的突變出現癲癇輕表型與癲癇重表型的概率無明顯差異,在N/C末端更容易出現癲癇輕表型,而在S5~S6區域出現重表型的概率高于輕表型,這可能與孔區特殊的功能相關。有文獻報道在S4、孔區、C末端等特殊功能處的突變可能會導致較重的癲癇表型,如早發性癲癇性腦病等[18]。曾琦等[22]報道在攜帶KCNQ2基因點突變的患兒中,智力運動發育正常的患兒多位于跨膜區外,而位于跨膜區S4、S5、S6和P環區的突變導致的表型均伴智力落后。KCNQ2引起的BFNE可分布在其蛋白各個結構域,而早發性癲癇腦病相關的突變多分布于S4、P環區域以及C端蛋白結合域等重要區域[23]。本研究錯義突變在S4區、N/C末端出現癲癇輕表型與重表型概率無明顯差異,需要更多的研究進一步證實其相關性。
4 小結
KCNQ2突變所引起的癲癇臨床表型輕重不一,其基因突變類型和突變位置均與臨床表型存在相關性。相比于無效突變,錯義突變更容易出現較重的癲癇表型;孔區突變所引起的癲癇的突變重表型更為多見,而C末端蛋白結合區的表型相對較輕。雖然本研究已經總結了常用數據庫的KCNQ2相關突變,但仍可能存在病例的遺漏情況。隨著KCNQ2突變致癲癇基因突變譜和疾病譜不斷豐富,突變與表型的相關性將進一步被揭示。
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國民間療法(2021年5期)2021-06-09 09:21:04
中國生殖健康(2020年2期)2021-01-18 02:51:26
家庭醫學(下半月)(2019年9期)2019-10-12 08:04:06
家庭醫學(下半月)(2019年8期)2019-09-25 09:02:00
小學生導刊(2018年13期)2018-06-29 03:49:00
飲食科學(2017年5期)2017-05-20 17:11:53
媽媽寶寶(2017年3期)2017-02-21 01:22:12
中國衛生標準管理(2015年8期)2015-01-26 18:08:35
西南軍醫(2015年4期)2015-01-23 01:19:30
