固相萃取-氣相色譜/質譜聯用法檢測茶葉中15種農藥殘留
2023-11-11 10:07:36馬淑青代飛飛聶丹丹
食品安全導刊 2023年27期
關鍵詞:標準
李 琳,馬淑青,代飛飛,聶丹丹
(濰坊市疾病預防控制中心,山東濰坊 261061)
山東省作為江北茶區之一,所產日照綠茶、嶗山綠茶、莒南茶、沂蒙綠茶等受到廣大消費者的歡迎。近年來,由于茶葉飲用安全問題,其多類農藥殘留的分析檢測成為行業關注的焦點[1-2]。考慮到茶葉基質復雜,建立了固相萃取[3]-氣相色譜/質譜聯用法[4-5](Gas Chromatography-Mass Spectrometry,GC/MS)測定茶葉中15種農藥殘留的分析方法。
1 材料與方法
1.1 材料與試劑
15種農藥標準品(100 μg·mL-1),均購自農業農村部環境保護科研監測所;乙腈、丙酮、正己烷和二氯甲烷,美國Fisher;氯化鈉(優級純)、無水硫酸鎂(分析純),國藥集團;Cleanert PC/NH2-SPE固相萃取柱(500 mg/500 mg/6 mL),美國Agela。
1.2 儀器與設備
手動固相萃取儀(北京萊伯泰科);ISQ單四極桿GC/MS聯用儀(美國賽默飛);N-EVAP氮吹儀(美國Organomation);渦旋振蕩器(德國Heidolph);LYNX400高速落地離心機(美國賽默飛);ARA520電子天平(奧豪斯)。
1.3 標準儲備溶液與標準溶液配制
將置于室溫平衡后的15種農殘標準溶液全部移入50 mL容量瓶中,用正己烷溶液(色譜級)少量多次清洗各標準溶液小瓶合并洗液至容量瓶中,定容至50 mL,配制成濃度為2.0 μg·mL-1的混合標準儲備液。再將標準儲備液稀釋成濃度為0.2 μg·mL-1、0.4 μg·mL-1、0.6 μg·mL-1、0.8 μg·mL-1和1.0 μg·mL-1的標準系列溶液。
1.4 實驗條件
1.4.1 色譜條件
色譜柱:TG-5MS(30 m×0.25 mm,0.25 μm);載氣:高純He;柱流量:1.0 mL·min-1;進樣口溫度:280 ℃;采用不分流進樣,進樣量:1 μL;程序升溫:40 ℃保持1 min,之后以30 ℃·min-1升溫至130 ℃,再以5 ℃·min-1升溫至250 ℃,最后以10 ℃·min-1升溫至300 ℃并保持5 min;接口溫度:280 ℃。
1.4.2 質譜條件
轟擊電子能量:70 eV;全掃描范圍:40~550 amu;采用全掃描模式和選擇離子監測模式采集信號;電離源溫度:230 ℃。
1.5 樣品提取與凈化
準確稱取1.0 g均勻粉碎的茶葉樣品于50 mL離心管中,加水10 mL渦旋混勻并浸泡30 min,再加入10 mL乙腈和1 g氯化鈉,渦旋振蕩0.5 min,待液液分層后加入4 g無水硫酸鈉,迅速旋上瓶蓋,振蕩渦旋1 min,10000 r·min-1離心5 min,取5.0 mL上清液于45 ℃水浴中氮吹至近干,10 mL丙酮分3次加入,提取液采用二氯甲烷(1∶1)洗脫樣品,洗脫液經過PC/NH2-SPE固相萃取柱凈化提取,收集于KD濃縮瓶中,氮吹至0.5 mL,待GC/MS分析測定。
2 結果與分析
2.1 色譜條件的優化
根據“相似相容”原理,選擇適合各待測組分化學性質的TG-5MS色譜柱分離15種農藥殘留;40 ℃保持1 min,以30 ℃·min-1升溫至130 ℃,以5 ℃·min-1升溫至250 ℃,再以10 ℃·min-1升溫至300 ℃保持5 min;進樣方式選擇不分流進樣。通過全掃描方式確定每種待測目標物的保留時間和定性定量離子,見表1。總離子流圖(見圖1)顯示,在TG-5MS色譜柱中15種農藥殘留能有效分離。
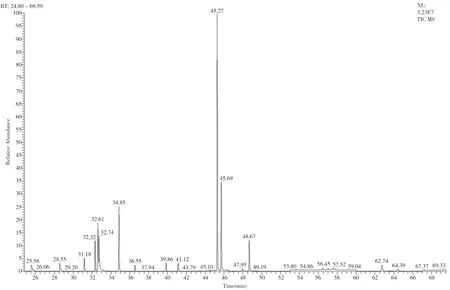
圖1 15種農藥的總離子流圖(TIC圖)
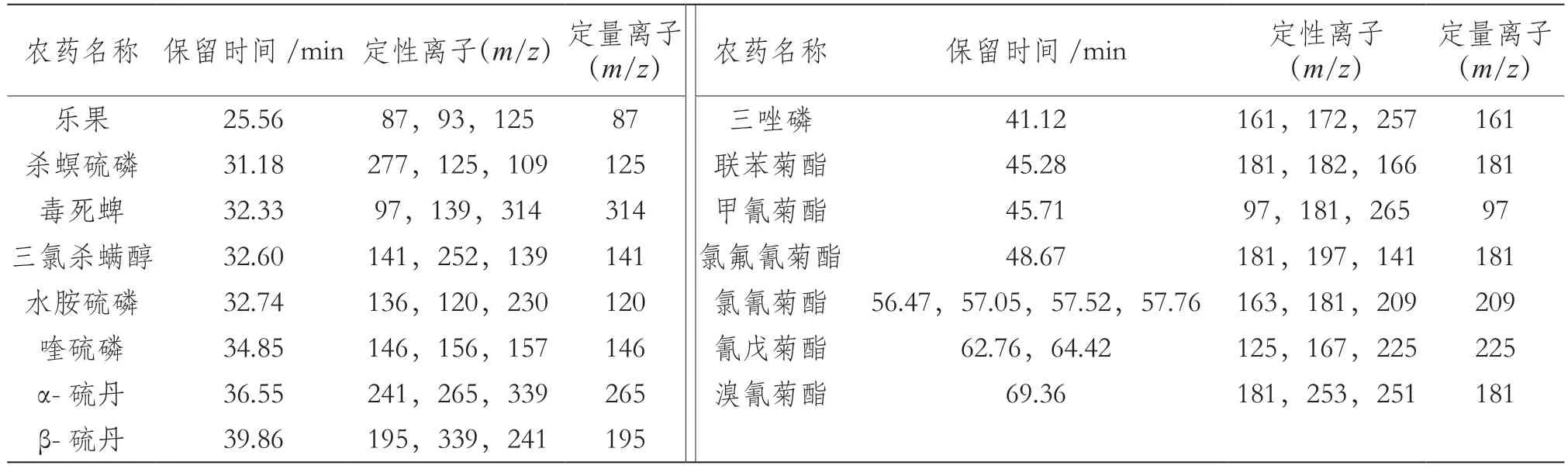
表1 15種農藥的保留時間和特征定性定量離子
2.2 方法的線性范圍、標準曲線和檢出限
混合農藥殘留標準系列配制5個濃度,濃度在0.2~1.0 μg·mL-1,以濃度為橫坐標、以峰面積為縱坐標繪制標準曲線并進行線性回歸分析。15種農藥標準溶液的回歸方程、相關系數、檢出限見表2。
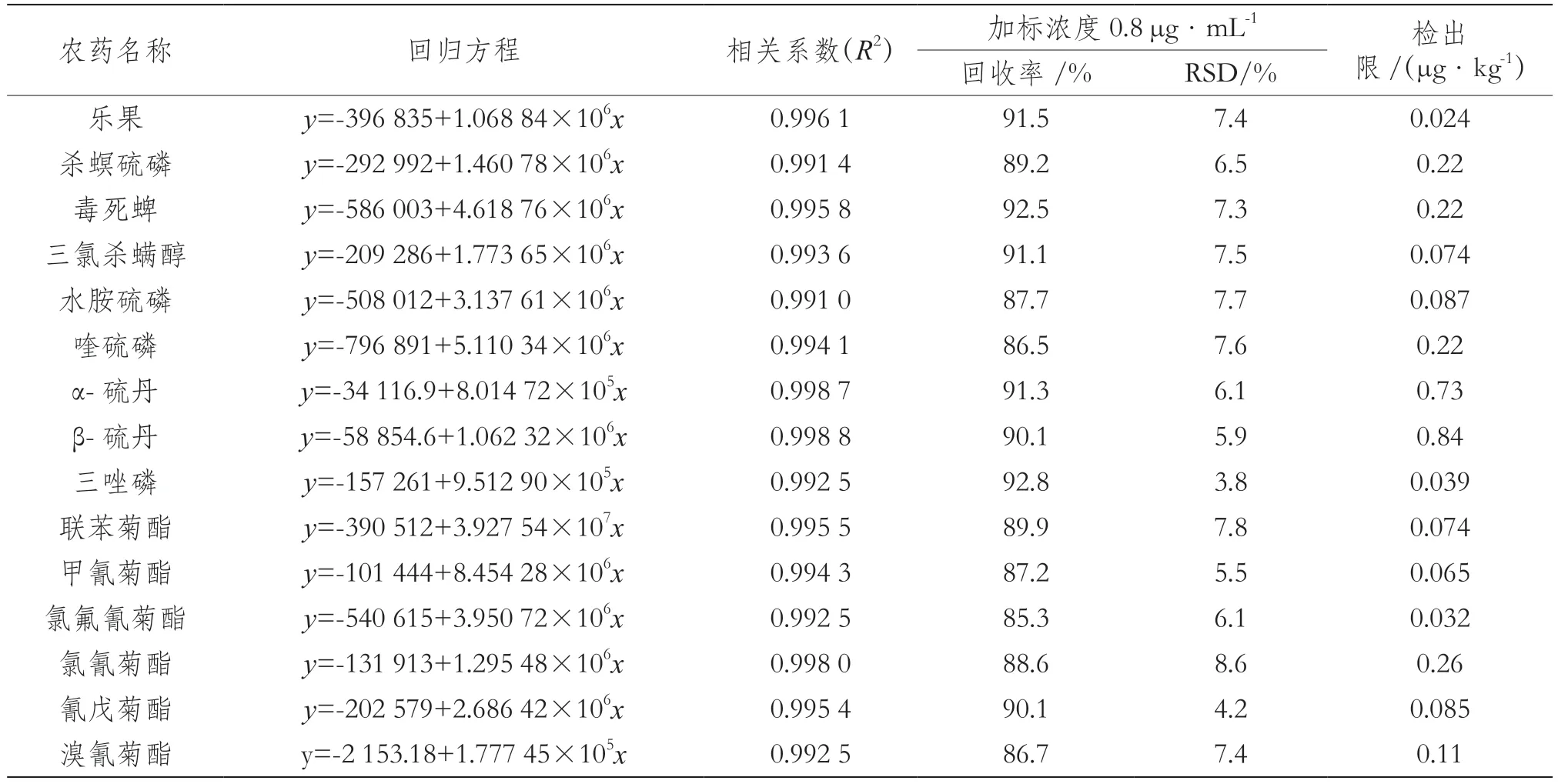
表2 15種農藥標準溶液的回歸方程、相關系數、回收率、精密度
2.3 方法的精密度與回收率
準確稱取1.0 g空白茶葉樣品6份,分別加入0.8 μg·mL-1的混合標準溶液,混勻后靜置1 h,按上述1.5試驗步驟提取凈化后上機測定,分別計算其加標回收率與相對標準偏差(Relative Standard Deviation,RSD),結果見表2。結果表明,方法的平均加標回收率為85.3%~92.8%,各待測組分相對標準偏差(n=6)為3.8%~8.6%,符合農藥殘留分析的要求。
2.4 樣品測定
用本文建立的方法對80份茶葉進行檢測,檢出農藥殘留46份,檢出率57.5%;超標樣品5份,超標率6.25%。
3 結論
本文建立了同時測定茶葉中15種常見農藥殘留的固相萃取-氣相色譜/質譜聯用分析方法,方法回收效果好,操作簡便快速,準確度與精密度均較高,適用于茶葉中多種農藥殘留的測定。
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
當代陜西(2019年8期)2019-05-09 02:22:48
上海建材(2019年1期)2019-04-25 06:30:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
家庭影院技術(2018年4期)2018-05-09 07:07:52
專用汽車(2016年4期)2016-03-01 04:13:43
質量與標準化(2015年9期)2015-12-31 11:41:40
中國質量與標準導報(2014年4期)2014-03-11 19:54:25
中國質量與標準導報(2014年10期)2014-02-28 22:25:47
中國質量與標準導報(2014年7期)2014-02-28 22:24:39
